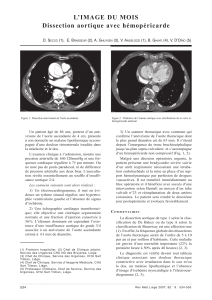
Signes diagnostiques et traitement d`une maladie de marfan ou

DOSSIER
© 2013 Elsevier Masson SAS. Tous droits réservés 23AMC pratique n°223 décembre 2013
MISE AU POINT
Le syndrome de Marfan est une patho-
logie génétique, en rapport généra-
lement avec une mutation dans le
gène FBN1, qui code pour une protéine de
la matrice extracellulaire, la fibrilline de
type 1. Ses limites nosologiques sont régu-
lièrement redéfinies (figures 1 et 2 pour
la dernière nosologie) du fait de la décou-
verte de pathologies proches qui corres-
pondent à des mutations dans d’autres
gènes (syndromes apparentés) [1]. Les
progrès réalisés au cours des 30 dernières
années ont permis de prolonger l’espé-
rance de vie des patients de 30 ans.
Le syndrome de Marfan
associe plusieurs signes
Des signes cardiovasculaires
Ils font toute la gravité pronostique de la
pathologie du fait du risque de dissection
aortique et de rupture.
La dilatation aortique prédomine au
niveau des sinus de Valsalva. Elle progresse
en moyenne de 0,5 mm/an dans l’ensemble
de la population Marfan (mais
certains patients se dilatent
moins vite que d’autres) et
peut se révéler tout au long de
la vie (figure 3). Ceci justifie
une surveillance des diamètres
aortiques tous les ans, tout au long de la vie,
même lorsque les diamètres sont dans les
valeurs normales. Cette surveillance est
généralement réalisée par échocardiographie
et on demande habituellement une confir-
mation du diamètre mesuré au moins une
fois par une autre technique (généralement
scanner) afin de s’assurer de sa fiabilité
(figure 4).
Sur le plan thérapeutique, on recommande
de limiter les efforts physiques lors desquels
la pression artérielle s’élève trop : éviter la
compétition, éviter les efforts isométriques
(dont l’archétype est l’haltérophilie mais
que l’on retrouve dans le basket, le
badminton, le hand-ball, le football…). Les
efforts d’endurance, comme par exemple la
natation, la course à pied, le vélo sans
compétition sont autorisés.
Il est également recommandé de mettre
en place un traitement bétabloquant chez
ces patients, avec l’idée que la diminution
de la fréquence cardiaque va limiter le
nombre de distensions brusques de la paroi
aortique et que la diminution de la force
de contraction du cœur va limiter la vitesse
de distension aortique (diminution du dp/
dt), ce qui va limiter le stress appliqué sur
la paroi aortique et de ce fait, ralentir la
dilatation et diminuer le risque de dissec-
tion. Ce traitement a été validé dans une
étude randomisée ancienne (figure 5). En
l’absence de tolérance des bétabloquants
(essentiellement asthme), on propose clas-
siquement un inhibiteur calcique qui a des
effets hémodynamiques similaires (brady-
cardie et inotrope négatif).
Des études sont en cours pour évaluer le
bénéfice d’un traitement par sartans, béné-
fice suspecté sur la base d’études chez la
souris génétiquement modifiée (Kl) [2]. Chez
G. Jondeau
- Centre national de référence pour le syndrome de Marfan et apparentés
- Equipe Insuffisance cardiaque
- Service de cardiologie, hôpitaux universitaires Paris Nord Val-de-Seine, hôpital Bichat, Paris
- INSERM U698
Signes diagnostiques et traitement
d’une maladie de Marfan ou apparentées
Mutation du gène FBN1
qui code pour
la fibrilline de type 1.

Signes diagnostiques et traitement d’une maladie de Marfan ou apparentées
MISE AU POINT
24
DOSSIER
AMC pratique n°223 décembre 2013
cette souris, de très fortes doses de losartan
ont permis la limitation de la dilatation de
l’aorte et le maintient de la structure histo-
logique de la paroi aortique. Il a donc été
proposé de tester l’efficacité du losartan chez
les patients qui présentent un syndrome de
Marfan. De nombreuses études sont en cours
chez l’homme. Les premiers résultats rappor-
tés chez l’homme sont encourageants, avec
une limitation de la vitesse de dilatation de
l’aorte chez les patients [3]. Cette première
étude porte sur de petits effectifs et on
espère que les autres études confirmeront
le bénéfice de cette molécule. Par son action
vasodilatatrice, elle pourrait diminuer l’onde
de rebond, voire baisser la pression artérielle
moyenne (mais les patients Marfan ne sont
pas hypertendus). L’hypothèse initiale selon
laquelle la molécule bloque l’anomalie qui
était proposée comme étant responsable
de la dilatation aortique (voie du TGF-béta)
semble moins probable aujourd’hui. On a
même des arguments pour penser que blo-
quer la voie du TGF-béta est délétère (par
exemple certaines des mutations en cause
chez les patients qui présentent des ané-
vrysmes diminuent ou bloquent la transmis-
sion du signal TGF-béta).
Le risque de dissection reste très limité tant
que le diamètre aortique reste en dessous de
50 mm, ou plus exactement, une attitude qui
consiste à remplacer l’aorte initiale lorsque
le diamètre atteint ou dépasse 50 mm est
associée à un risque très faible de dissection
aortique (5 % pour 100 ans de suivi) [4]. On
considère donc maintenant que 50 mm est
le seuil opératoire, accepté dans les dernières
recommandations [5]. De plus en plus souvent,
la chirurgie aortique préventive permet de
conserver les valves natives (plastie de l’aorte
ascendante) et le risque opératoire est minimal
Figure 1. Critères diagnostiques proposés pour le syndrome de Marfan lors de la dernière
réunion consensus.
Ao : dilatation aortique
Z : Z score
FBN1
: mutation dans le gène
FBN1
MFS : syndrome de Marfan
EL : ectopie du cristallin
Syst : score systémique (voir
figure 5
)
Pb Ao : problème aortique
ELS : syndrome d’ectopie du cristallin
MASS : syndrome MASS (Mitral Aorta Skelet Skin)
PVM : prolapsus valvulaire mitral
Figure 2. Calcul du score systémique utilisé dans la dernière classification proposée.
Figure 3. Mode de mesure du diamètre aortique maximal,
sur la vue parasternale grand axe, en prenant la paroi
antérieure et non la paroi postérieure, en télédiastole
(sur le QRS).

MISE AU POINT
G. Jondeau
25
DOSSIER
AMC pratique n°223 décembre 2013
chez des patients jeunes, dont la fonction ven-
triculaire gauche est normale. Différentes tech-
niques sont proposées, dont le résultat dépend
notamment de l’expérience du chirurgien dans
ce type d’intervention (cf. article de M. Kirsch et
U. Hvass).
La prise en charge de la grossesse pose
des problèmes particuliers : schématique-
ment, le risque de dissection est considéré
comme augmentant à partir de 40 mm et
la chirurgie prophylactique peut être pro-
posée à partir d’un diamètre de 45 mm [5].
Se pose également le problème du mode de
délivrance, voie basse en dessous de 40 mm
et généralement césarienne au dessus. Une
surveillance échocardiographique étroite
doit être mise en place avec une échogra-
phie à 3 mois, 6 mois, puis tous les mois au
cours du troisième trimestre et au décours
de l’accouchement.
D’autres conditions sont considérées comme
à risque et peuvent justifier d’une chirurgie
plus précoce : antécédent de dissection dans
la famille à un faible diamètre, progression
rapide (de 3 mm en un an d’après les der-
nières recommandations – seuil arbitraire –,
à vérifier avec deux techniques différentes,
en revoyant les examens simultanément).
Même si le risque principal se situe sur l’aorte
ascendante, l’atteinte aortique est diffuse, si
bien qu’il existe également un risque de dis-
section de l’aorte descendante [6]. Cette dis-
section peut survenir alors que le diamètre
de l’aorte ascendante est normal et ceci jus-
tifie de proposer le traitement bétabloquant
et de proscrire les sports violents quelque soit
le diamètre de l’aorte ascendante. De même,
cette attitude est à poursuivre après rempla-
cement de l’aorte ascendante.
Le prolapsus valvulaire mitral est fréquent,
souvent bivalvulaire équilibré avec une fuite
modérée qui nécessite rarement la chirurgie
(figure 4). La chirurgie est souvent une
plastie, parfois délicate du fait de l’étendue
du prolapsus, de l’importance de la dilatation
de l’anneau mitral.
Des signes squelettiques
Ils sont responsables de l’aspect classique
(figure 6).
La croissance excessive des os longs peut
entraîner pectus excavatum ou recurva-
tum, arachnodactylie, scoliose. Les patients
peuvent également présenter des pieds
plats. Les conséquences en sont essentielle-
ment fonctionnelles avec des douleurs qu’il
est difficile de soulager, des problèmes
esthétiques qui peuvent rendre les patients
demandeurs de chirurgie esthétique. La sco-
liose et les pieds plats peuvent justifier une
intervention orthopédique.
Des vergetures peuvent être présentes sur
tout le corps et la localisation sur le devant
des épaules est évocatrice.
Figure 4. Apparition des signes cardiologiques en fonction de l’âge des patients.
Figure 5. Bénéfice du traitement bétabloquant
Aortic ratio : rapport du diamètre mesuré sur le diamètre théorique
IAo : insuffisance aortique

Signes diagnostiques et traitement d’une maladie de Marfan ou apparentées
MISE AU POINT
26
DOSSIER
AMC pratique n°223 décembre 2013
Les vertèbres peuvent être déformées par une
ectasie du sac dural (figure 6), visible essen-
tiellement au scanner ou à l’IRM. Il n’y a géné-
ralement pas de symptôme associé (rarement
céphalées) ni de traitement à prévoir.
L’ectopie du cristallin est un signe majeur
(comme l’anévrysme aortique), est souvent
supérieure et temporale, et souvent
incomplète, justifiant une dilatation
soigneuse pour sa recherche (figure 6). Les
autres signes ophtalmologiques sont cornées
plates, myopie secondaire à une longueur
axiale augmentée (la chirurgie de la myopie
n’est pas indiquée).
Un pneumothorax peut également être
observé. C’est en fait assez rare dans cette
population.
Origine génétique
Le fait fondamental est l’origine génétique
de la pathologie, qui est transmise selon le
mode dominant autosomique. Un des deux
parents est atteint sauf si la mutation appa-
raît pour la première fois (néomutation), ce
qui est le cas chez 25 % des patients, et un
patient atteint a une chance sur deux de
transmettre la pathologie sans préférence
de sexe. Ceci justifie de rechercher la pré-
sence des signes cliniques chez les apparen-
tés d’un patient atteint, notamment dans
le but de dépister un anévrysme aortique
avant qu’une dissection ne survienne.
Ainsi, la prise en charge d’un patient sus-
pect de présenter le syndrome de Marfan
doit comprendre un examen ophtalmolo-
gique, un examen cardiologique avec écho-
cardiographie, et un examen systémique à
la recherche des autres signes ; ceci est réa-
lisé au mieux dans le centre de référence ou
les centres de compétence, mis en place par
le plan maladies rares.
La réalisation d’une étude de biologie
moléculaire à la recherche d’une mutation
dans le gène FBN1 est réalisée de plus en
plus souvent lorsque les signes évocateurs
le justifient. Elle a l’intérêt de confirmer
le diagnostic dans les formes douteuses et
de faciliter l’enquête familiale. Elle reste
néanmoins chère et soumise au filtre du
laboratoire de biologie moléculaire, du fait
de sa difficulté (le gène est long et chaque
famille a une mutation qui lui est propre),
et du faible rendement de la recherche de
mutation en l’absence d’atteinte ophtal-
mologique. La demande excessive conduit
à des délais d’attente longs, qui devraient
s’améliorer avec les progrès des techniques
de biologie moléculaire.
Traitement
Une fois le diagnostic porté, le patient doit
éviter les sports violents (risque de luxation
du cristallin par des sports de contact et
risque de favoriser la dilatation aortique en
cas d’élévation tensionnelle lors d’un effort
brusque intense), un traitement bétablo-
quant entrepris et s’il n’est pas toléré, un
traitement par inhibiteur calcique ralentis-
seur ou un sartan. Une surveillance écho-
cardiographique annuelle est proposée
[5]. Ceci a permis d’obtenir des risques de
dissection très faibles aujourd’hui [4] et de
réaliser plus souvent une chirurgie préven-
Figure 6. Aspect général avec la scoliose, le
pectus
, etc.
En haut à droite : ectopie du cristallin.
En bas à droite : ectasie durale.

MISE AU POINT
G. Jondeau
27
DOSSIER
AMC pratique n°223 décembre 2013
tive de l’aorte qu’une chirurgie d’urgence
pour dissection (figure 7).
Le traitement de la luxation du cristallin
repose sur son ablation justifiée en cas de
conséquence fonctionnelle importante et
pose le problème de la mise en place d’im-
plant qui est parfois difficile et toujours non
recommandée chez les enfants. En l’absence
d’implant, les lunettes sont nécessaires.
Le traitement rhumatologique n’est pas
spécifique (traitement de la scoliose, traite-
ment des pieds plats, des douleurs…).
Enfin, du fait de l’origine génétique de la
pathologie, il est fondamental de réaliser
une enquête familiale pour dépister les
parents atteints qui ne le sauraient pas,
avant qu’une dissection aortique ne révèle
chez eux la maladie.
Les syndromes apparentés
Les progrès réalisés ces dernières années
reposent également sur le fait que de nou-
veaux syndromes aient pu être reconnus,
dont le pronostic peut différer ou non du
syndrome de Marfan classique, notamment
parmi les formes familiales d’anévrysmes.
Certains sont associés à des signes systé-
miques, d’autres non [7].
L’importance des signes rhumatologiques,
la survenue précoce d’une arthrose doit
faire évoquer une mutation dans le gène
SMAD3 (osteo-arthritis syndrome), la pré-
sence d’une peau fine translucide, une
luette bifide, un hypertélorisme : une muta-
tion dans le gène TGFBR1 ou TGFBR2 (Loeys
Dietz syndrome) ; et un livedo racemosa :
une mutation dans le gène ACTA2.
La persistance d’un canal artériel fait évo-
quer une mutation dans le gène MYH11.
Des tableaux de Marfan classique y compris
correspondant aux critères récents, avec signes
squelettiques plus ou moins nets, l’absence de
signe ophtalmologique franc peuvent se voir
chez les porteurs de mutation dans le gène
TGFBR1, TGFBR2, SMAD3 et TGFB2.
Mais toutes ces mutations peuvent ne se tra-
duire que par un anévrysme de l’aorte ascen-
dante isolé (mais génétique, donc familial).
Quel que soit le type de mutation en cause,
le diagnostic de syndrome de Marfan ou
d’une forme apparentée conduit à la limi-
tation des sports, à la prescription d’un trai-
tement bétabloquant et à une surveillance
échocardiographique annuelle. Mais cer-
taines mutations dans ces nouveaux gènes
peuvent s’accompagner d’un risque d’ané-
vrysme cérébral, ce qui justifie un bilan vas-
culaire cérébral initial (IRM, voire scanner).
Certaines mutations s’accompagnent égale-
ment d’un risque de dilatation et de dissec-
tions d’artères périphériques, qui justifie la
réalisation d’une opacification de toutes les
artères au moins une fois chez les patients
porteurs de ces mutations (généralement
un scanner de l’ensemble des vaisseaux). Il
s’agit de formes génétiques de dilatation
ou dissections d’artères périphériques, qui
sont donc soupçonnées soit devant l’exis-
tence de signes extra-aortiques évocateurs
(arthrose précoce, luette bifide, peau fine et
transparente, hypertélorisme…), soit devant
la pathologie aortique et son caractère
familial. Ce dernier point souligne encore
la nécessité de faire une enquête familiale
pour dépister les apparentés qui présen-
tent un anévrysme de l’aorte ascendante,
Figure 7. Evolution de la prise en charge des patients illustrée par la modification des
indications des chirurgies aortiques (histogramme supérieur) et le type d’intervention réalisée
(histogramme inférieur).
 6
6
1
/
6
100%