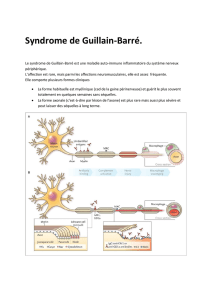
Le syndrome de Guillain-Barré

Revue flash
Neurologie.com 2009 ; 1 (1) : 19-22
DOI : 10.1684/NTO 2008 02 neurologie.com |vol. 1 n°1 |mars 2009 19
webneurologie.com
L’évaluation sur
Le syndrome
de Guillain-Barré
Guillain-Barré syndrome
Gérard Said
Fédération de neurologie,
Hôpital de la Pitié-Salpêtrière,
Paris
<dr-gs@hotmail.fr>
Mots clés
syndrome de Guillain Barré,
polyradiculonévrites
Key words
acute polyneuritis,
demyelinating poly neuro pathy,
neuropathology
Pour la pratique on retiendra
Le syndrome de Guillain-Barré (SGB) résulte d’une réponse immunitaire à une agression le plus souvent virale. Le diagnostic repose
sur des critères cliniques, sur les modifications du liquide céphalo-rachidien et sur les anomalies électrophysiologiques. Le
traitement symptomatique des défaillances respiratoires et des troubles engendrés par les paralysies en a considérablement réduit
la mortalité, mais des séquelles sensitivomotrices ne sont pas rares. Les immunoglobulines intraveineuses administrées
précocement ont supplanté les échanges plasmatiques, mais restent d’une efficacité modérée.
Abstract
Guillain-Barré syndrome likely results from an immune response to a viral infection in most cases, without any immunological marker.
The diagnosis rests on clinical manifestations, and CSF and electrophysiological changes. Symptomatic treatment of respiratory
failure and complications of prolonged paralysis have strikingly reduced the mortality of the syndrome, but residual deficits are com-
mon. Early administration of high doses intravenous immunoglobulins is currently preferred to plasma exchanges, but their efficacy
remains modest.
Décrit par Guillain, Barré et Strohl,
en 1916 [1], le syndrome de
Guillain-Barré (SGB) est considéré
comme le résultat d’une réponse
immunitaire à une agression le
plus souvent virale. Malheu -
reusement, le ou les antigène(s) en cause n’ayant
toujours pas été identifié(s), il n’y a pas de marqueur
biologique de l’affection, donc le diagnostic repose
sur des critères cliniques, sur les modifications du
liquide céphalo-rachidien, et les anomalies de la
conduction nerveuse. Les traitements modernes, et
en particulier les soins intensifs en cas de besoin,
ont permis de réduire considérablement la
mortalité du syndrome, dont les séquelles
sensitivomotrices sont en fait assez fréquentes.
ÉPIDÉMIOLOGIE
Le nombre de nouveaux cas est en moyenne d’un
par an pour 100 000 habitants. Chez deux tiers des
patients on note un antécédent infectieux des voies
aériennes supérieures, ou digestives (pharyngite ou
diarrhée), une à trois semaines avant les premiers
symptômes de neuropathie. Parmi les agents
infectieux les plus souvent identifiés, on rencontre
les virus de la famille des herpes virus
(cytomégalovirus, virus d’Epstein-Barr, plus
rarement zona), le virus de l’immunodéficience
humaine (VIH), le mycoplasme surtout chez l’enfant
et l’adulte jeune, et le
Campylobacter jejuni
responsable d’entérites aiguës bactériennes. Dans
moins de 2 à 3% des SGB, l’événement prodromique
consiste en un acte chirurgical, une sérothérapie
ou une vaccination. Certaines affections
dysimmunitaires telles la maladie de Hodgkin, les
lymphomes non hodgkiniens ou le lupus
érythémateux aigu disséminé peuvent s’associer ou
se révéler par un SGB.
LÉSIONS NERVEUSES
ET PHYSIOPATHOLOGIE DU SGB
La lésion élémentaire du SGB est une
démyélinisation aiguë prédominant sur la partie
proximale sous-arachnoïdienne du SNP et touchant
éventuellement de façon asymétrique et
multifocale les nerfs périphériques [2]. Les lésions
associent infiltrats lymphocytaires et
macrophagiques et démyélinisations segmentaires
des fibres nerveuses périphériques
(figures 1 et 2)
.
À la démyélinisation complète de l’espace
internodal succède la remyélinisation réparatrice.
L’axone classiquement respecté peut être lésé en
cas de lésion inflammatoire intense. Ces lésions
axonales secondaires rendent compte des
amyotrophies distales et des séquelles.
La remyélinisation devient évidente au 15ejour
après une démyélinisation aiguë expérimentale.
LE SYNDROME DE GUILLAIN-BARRÉ
Dans le syndrome de Guillain-Barré, le patient perd progressivement
la force musculaire des jambes, des bras, de la face, et parfois même
la force d’avaler et de respirer. Ainsi,
tous les muscles du corps sont
affaiblis. Ce manque de force peut...
webneurologie.com
La suite sur
Résumé pour les patients
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 26/05/2017.

20 neurologie.com |vol. 1 n°1 |mars 2009
Les démyélinisations focales sont à l’origine de blocs de
conduction et de déficits neurologiques dans le territoire
correspondant. La remyélinisation est un processus de
réparation rapide qui fait disparaître le bloc de conduction
et le déficit neurologique qui en dépendait. Dans tous les cas,
le SGB est limité dans le temps, pratiquement toujours
monophasique, mais la récupération des déficits est variable.
CLINIQUE
Forme typique
▀Aspects cliniques
Des troubles sensitifs subjectifs, comme des
engourdissements distaux, souvent asymétriques, et des
myalgies surviennent dans près de la moitié des cas,
souvent avant l’installation des déficits moteurs qui
caractérisent le syndrome [3].
L’intensité du déficit moteur est variable. Les parésies et
paralysies s’installent rapidement faisant toute la gravité
potentielle du SGB. Le déficit moteur atteint son maximum en
moins de deux semaines dans 80% des cas. Au terme de cette
phase de progression des déficits, on entre dans la phase dite
de plateau de durée variable, pouvant aller de quelques jours
à plusieurs mois, qui va précéder la récupération spontanée.
Les déficits touchent à la fois la partie proximale et distale
des membres dans la moitié des cas. Ils prédominent à la
partie distale dans un tiers des cas, et à la partie proximale
chez les autres. Les réflexes ostéotendineux sont abolis dans
le territoire correspondant, mais parfois avec retard. Leur
conservation ne doit pas faire récuser le diagnostic.
L’amyotrophie est tardive. Sa précocité est un argument de
mauvais pronostic fonctionnel car elle témoigne d’une
atteinte axonale. L’intensité des paralysies est variable,
allant d’une simple faiblesse des membres inférieurs gênant
la marche et la montée des escaliers, à la tétraplégie
complète avec paralysie des muscles respiratoires nécessitant
une intubation et une ventilation mécanique d’urgence,
paralysie des muscles de la déglutition, diplégie faciale,
ophtalmoplégie et aréflexie diffuse.
Le nerf facial est affecté dans la moitié des cas, souvent de
manière bilatérale et symétrique. L’atteinte des nerfs du
carrefour aérodigestif expose à des troubles de la déglutition
dans un quart des cas environ. Une ophtalmoplégie externe
est relativement rare.
Au cours de l’évolution, il est fréquent de noter une
hypoesthésie distale avec diminution de la sensibilité
vibratoire, des erreurs au sens de position des orteils,
astéréognosie et hypoesthésie tactile.
Certains signes inhabituels peuvent s’observer comme un
œdème papillaire ou la survenue d’un tremblement
d’attitude, en rapport avec l’existence d’un déficit moteur
mineur agravé par la présence de troubles proprioceptifs.
Des cas de SGB ont été rapportés lors de séroconversion VIH,
en association à un tableau de virose aiguë avec fièvre,
diarrhée, exanthème, polyadénopathie avec syndrome
mononucléosique. La séroconversion était observée dans les
semaines suivantes.
▀Liquide céphalo-rachidien (LCR)
Une hyperprotéinorachie supérieure à 0,45 g/L, pouvant
atteindre des taux de plus de 10 g/L, sans réaction cellulaire
est caractéristique, mais fait défaut dans 15% des cas, même
en répétant les vérifications du LCR. La persistance et
l’importance de l’hyperprotéinorachie n’ont pas de
signification pronostique. On observe parfois la présence de
quelques lymphocytes dans le LCR. Chez les patients porteurs
du VIH, la réaction cellulaire dans le LCR est habituelle.
Variantes cliniques
Le SGB est en fait assez polymorphe.
▀Syndrome de Miller-Fisher
Il se définit par la triade ophtalmoplégie, ataxie, aréflexie
ostéotendineuse et représente 5% des SGB [4]. Des paresthésies
distales et une faiblesse modérée des muscles proximaux et
de la déglutition peuvent parfois s’y associer. Cette forme
clinique peut évoluer vers un SGB avec un déficit moteur
généralisé et une paralysie des muscles respiratoires, mais
elle reste le plus souvent bénigne. Les études
électrophysiologiques de ce syndrome ont toutes démontré
des signes de neuropathie périphérique. L’ophtalmoplégie est
souvent associée à une élévation non spécifique des anticorps
anti-GQ1b [5].
▀Formes purement motrices
Elles représentent environ 3% des SGB. Les lésions
prédominent sur les racines antérieures, ainsi que l’ont
Figure 1. Coupe semi-fine d’une biopsie de nerf musculo-cutané d’un patient
présentant un syndrome de Guillain-Barré en phase de plateau très prolongée.
Notez les nombreuses fibres démyélinisées (flèches). Échelle: 20 µm.
Figure 2. Microscopie électronique. Syndrome de Guillain-Barré atypique. Biopsie
de nerf montrant un axone démyélinisé entouré de nombreuses cellules
macrophages (M).
S: cellule de Schwann; Ax: axone démyélinisé.
M
Ax
S
2µ
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 26/05/2017.

neurologie.com |vol. 1 n°1 |mars 2009 21
confirmé les rares cas anatomiques. L’examen électrique peut
montrer des modifications caractéristiques des conductions
nerveuses, parfois des blocs de conduction. Un ralentissement
de la vitesse de conduction nerveuse peut s’observer sur les
nerfs sensitifs, même en l’absence d’atteinte clinique. C’est
dans ce sous-groupe que les formes motrices axonales aiguës,
dont la variabilité évolutive reflète celles du SGB, ont été
individualisées.
▀Formes purement sensitives
Elles sont rares et se caractérisent par des paresthésies
distales associées à un engourdissement et une aréflexie
ostéotendineuse alors qu’il y a peu ou pas de faiblesse
musculaire pendant la durée de la maladie.
▀Pandysautonomie aiguë idiopathique
C’est une forme très rare avec fatigue, vomissements, douleurs
abdominales, troubles du transit, hypotension orthostatique
avec parfois syncope et pouls fixe, impuissance, troubles de
la sudation, de la salivation, et anomalies pupillaires [6]. Les
réflexes ostéotendineux sont diminués ou absents. L’évolution
est prolongée sur plusieurs mois, voire des années. Cette forme
doit être différenciée des perturbations végétatives transitoires
des formes sévères de SGB typiques par ailleurs.
▀Formes axonales de SGB
Les formes graves avec lésions axonales prédominantes
représentent une extrémité du spectre du SGB, l’autre
extrémité étant représentée par les formes démyélinisantes
pures, de bien meilleur pronostic, avec entre les deux, des
formes plus ou moins graves selon la proportion de lésions
axonales.
▀Manifestations neurovégétatives et générales au cours de
l’évolution du SGB
Les manifestations végétatives sont nombreuses et
polymorphes, mais le plus souvent bénignes avec labilité
tensionnelle, et parfois poussées hypertensives préoccupantes.
Les troubles vésico-sphinctériens ont été notés dans près d’un
quart des cas. Ils régressent en une quinzaine de jours.
L’incontinence urinaire est plus rare et résulte le plus
souvent de mictions par regorgement. D’autres troubles
végétatifs sont également rencontrés de façon transitoire.
La plupart des complications générales que l’on rencontre dans
l’évolution d’un SGB sont en rapport avec l’immobilisation
prolongée, avec en particulier un risque d’embolie pulmonaire,
dont la prévention sera un souci constant.
ÉLECTROPHYSIOLOGIE
Les anomalies les plus évocatrices du SGB sont la baisse des
vitesses de conduction nerveuse, l’augmentation de la latence
de l’onde F qui témoigne d’une démyélinisation proximale,
l’allongement des latences distales, et la présence de blocs de
conduction sur les troncs nerveux, en dehors des zones de
compression habituelles [7]. L’abaissement des potentiels
d’action moteurs et sensitifs a une valeur pronostique, car elle
témoigne de la perte axonale associée. La répétition des
examens électriques n’est pas utile. Il arrive que toutes les
conductions nerveuses restent normales sans que cela ne
remette en cause le diagnostic.
ÉVOLUTION
Forme habituelle
Le SGB est caractérisé par une évolution en trois phases: phase
d’installation des déficits, puis, phase en plateau de durée
variable, et enfin, phase de régression de durée également
variable.
La première phase ascendante marquée par l’aggravation
progressive du déficit moteur et des paresthésies dure
généralement 11 à 12 jours en moyenne, mais peut être
beaucoup plus rapide avec un risque de défaillance respiratoire
précoce.
La phase de plateau, pendant laquelle les déficits restent
stationnaires, lui succède. Elle dure de quelques jours à
plusieurs mois.
Une phase de récupération est plus ou moins longue. Certains
patients guérissent complètement, mais 10% gardent des
séquelles plus ou moins lourdes. La mortalité actuelle est de
l’ordre de 3%.
Formes évolutives atypiques
Des rechutes, après guérison complète de la première
poussée, sont exceptionnelles. L’intervalle entre les deux
poussées est variable. Un SGB marque exceptionnellement le
début d’une polyradiculonévrite chronique, dont il existe des
formes lentement progressives, des formes évoluant par
paliers, des formes monophasiques lentes ou à rechutes, et
des formes progressives évoluant par poussées.
Facteurs pronostiques
Plus l’atteinte axonale associée à la démyélinisation est
importante, plus la récupération sera lente, et pourra laisser
des séquelles motrices ou douloureuses. On comprend le
caractère péjoratif d’une amyotrophie précoce, de signes
électriques importants de dénervation, d’un effondrement des
potentiels d’action, tous ces signes témoignant de l’intensité
de la perte axonale associée.
DIAGNOSTIC DIFFÉRENTIEL
Les difficultés diagnostiques rencontrées au cours des
différentes formes de SGB sont très variables.
Déficit moteur pur sans modification du LCR
Il faut exclure une affection musculaire aiguë acquise
comme une polymyosite ou une rhabdomyolyse aiguë par les
dosages d’enzymes musculaires, ou une paralysie
hypokaliémique par l’ionogramme sanguin. Une anomalie
de la jonction neuromusculaire: essentiellement une
myasthénie par un test pharmacologique à l’edophronium
et les dosages d’anticorps. Le botulisme peut poser des
problèmes difficiles quand les paralysies oculomotrices
sont au premier plan.
LCR normal ou hyperprotéinorachie modérée
Une polynévrite alcoolique aiguë ou une polyradiculonévrite
de réanimation peuvent se discuter ; mais c’est la porphyrie
aiguë intermittente qui est probablement l’affection la plus
trompeuse. Lorsque les antécédents familiaux sont connus,
le diagnostic est évident, mais lorsqu’ils ne le sont pas il faut
y penser devant une atteinte multifocale axonale,
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 26/05/2017.

22 neurologie.com |vol. 1 n°1 |mars 2009
symétrique ou asymétrique,
a fortiori
si on peut retrouver
la notion d’urines fonçant à la lumière, de douleurs
abdominales, et de prise médicamenteuse, barbiturique en
particulier.
En l’absence de signes pyramidaux francs
Une quadriplégie rapidement progressive par lésion aiguë ou
subaiguë mécanique, ischémique, hémorragique ou
inflammatoire de la moelle cervicale peuvent se discuter et
faire réaliser une IRM en urgence.
En présence d’une réaction cellulaire dans le LCR
Ne pas oublier que la poliomyélite antérieure aiguë existe
toujours, associant un déficit moteur périphérique subaigu
à une méningite lymphocytaire, avec dissociation
albuminocytologique tardive, contrairement au SGB dans
lequel cette dissociation est précoce.
Dans la maladie de Lyme, l’anamnèse, les signes cutanés, la
réaction lymphoplasmocytaire dans le LCR et la sérologie
permettront le diagnostic.
Les infiltrations malignes des méninges et des racines
peuvent en imposer pour une forme subaiguë de
polyradiculonévrite. L’identification de cellules malignes
dans le LCR et les prises de contraste méningées et radiculaires
à l’IRM aideront au diagnostic.
Les neuropathies des patients de réanimation surviennent au
cours de défaillances polyviscérales chez des sujets infectés
le plus souvent, avec défaillance respiratoire ayant reçu des
curarisants, difficulté à sevrer les patients du ventilateur [8].
Elles associent un déficit moteur des quatre membres,
d’intensité variable, axonal, longueur-dépendant. Le LCR est
subnormal.
TRAITEMENT
Traitement symptomatique
Il doit assurer la prévention et le traitement des complications
éventuelles des troubles de la déglutition, de la ventilation,
des troubles sphinctériens et des complications de décubitus
et la prévention des phlébites et des embolies pulmonaires
par héparinothérapie. Les douleurs devront être contrôlées
par des antalgiques. Les patients présentant des troubles
moteurs doivent être suivis en service spécialisé. Les ulcères
de stress et les gastrites érosives sont relativement fréquents.
Un soutien psychologique est indispensable chez ces patients
alités pour des semaines.
Le traitement du déficit moteur et la prévention des
rétractions tendineuses doivent se faire très précocement.
Immunothérapie dans le SGB
Depuis quelques années, des traitements de fortes doses
d’immunoglobulines par voie veineuse (IgIV) (0,4 g/kg/j, cinq
jours de suite) ont été préconisés. Ce traitement semble donner
des résultats supérieurs à ceux des échanges plasmatiques [9].
En pratique, nous recommandons ces traitements
immunomodulateurs essentiellement chez les patients en
phase de progression des paralysies.
Le pronostic du SGB, qui représente une neuropathie
démyélinisante aiguë ou subaiguë d’origine probablement
auto-immune, a été considérablement amélioré par les
méthodes de réanimation modernes, et de façon plus
modeste, par les traitements immunomodulateurs.
POINTS FORTS
• Le syndrome de Guillain-Barré est une polyneuropathie
démyélinisante.
• Son évolution est généralement monophasique.
• Il peut entraîner une quadriplégie flasque rapide avec
troubles respiratoires.
• La régression spontanée en quelques semaines est
l’évolution la plus fréquente.
• Plus l’atteinte axonale associée est importante, plus il y a de
risque d’évolution prolongée et de séquelles.
Références
1. Guillain G, Barré JA, Strohl A. Sur un
syndrome de radiculonévrite avec
hyperalbuminose du liquide céphalo-
rachidien sans réaction cellulaire. Bull
Mem Soc Med Hop Paris, 1916 : 1462-70.
2. Asbury AK, Arnason BG, Adams RD.
The inflammatory lesions in idiopathic
polyneuritis. Medicine 1969 ; 48 : 173-215.
3. Ropper AH, Wijdicks EFM, Truax BT.
Guillain Barré syndrome in Contempo-
rary Neurology Series. In : Plum F, ed.
Philadelphie : FA Davis Company, 1991.
4. Fisher M. An usnusual variant of acute
idiopathic polyneuritis (syndrome of
ophthal moplegia, ataxia and areflexia).
N Engl J Med 1956; 255 : 57-65.
5. Odaka M, Yuki N, Hirata K. Anti-GQ1b
IgG antibody syndrome : clinical and
immunological range. J Neurol Neuro-
surg Psychiatry 2001 ; 70 : 50-5.
6. Young RR, Asbury AK, Corbett JL,
Adams RD. Pure pandysautonomia with
recovery. Brain 1975; 98 : 613-36.
7. Brown WF, Feasby TE. Conduction
block and denervation in Guillain-Barré
polyneuropathy. Brain 1984; 107 : 219-39.
8. Bolton CF, Laverty DA, Brown JD, et al.
Critically ill polyneuropathy : electro-
physiological studies and differentiation
from Guillain-Barré syndrome. J. Neurol.
Neurosurg. Psychiatry 1986 ; 49 : 563-73.
9. Van der Meché FGA, Scmitz PIM,
Dutch Guillain-Barré Study Group. A ran-
domized trial comparing intravenous
immune globuline and plasma exchange
in Guillain Barré syndrome. N Engl Med
1992 ; 326 : 1123-9.
+
Et toujours
sur
www.
webneurologie.com
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 26/05/2017.
1
/
4
100%