GB_TALBI


Dr N.TALBI
20/02/2015
8ème Journée Pédiatrique de Mascara8ème Journée Pédiatrique de Mascara8ème Journée Pédiatrique de Mascara

Introduction
Le syndrome de Guillain, Barré reste toujours d’actualité, les progrès
scientifiques ont permis de le faire évoluer dans quatre directions :
La discussion nosologique et les définitions diagnostiques.
Les connaissances épidémiologiques et pronostiques.
Les aspects physiopathologiques et électro physiologiques
La stratégie thérapeutique.

Définition
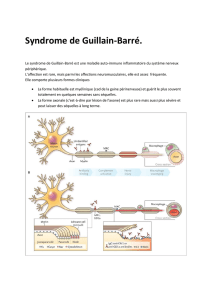
Le syndrome de Guillain-Barré(SGB) ou syndrome de Guillain-Barré-Strohl est
une maladie auto-immune inflammatoire acquise du système nerveux périphérique.
On l'appelle également :
polyneuropathie aiguë inflammatoire démyélinisante,
polyradiculonévrite aiguë idiopathique,
polynévrite aiguë idiopathique
paralysie ascendante de Landry.
Il se caractérise principalement par :
- un déficit moteur bilatéral, symétrique, ascendant, avec abolition des ROT
- des troubles sensitifs,
- une dissociation albumino-cytologique dans le LCR.

Historique
En 1859, le médecin français Jean Landry a décrit pour la
première fois ce syndrome.
En 1916, George Guillain, Jean Alexandre Barré et André
Strohl ont diagnostiqué deux soldats d’une paralysie généralisée
transitoire qui avaient dans la liquide céphalo-rachidien une
concentration augmentée des protéines, avec un tissu cellulaire
normal.
Cette découverte a été la clef du diagnostic de ce syndrome.
Jean Landry
George Charles Guillain André Strohl Jean-Alexandre Barré
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
1
/
40
100%