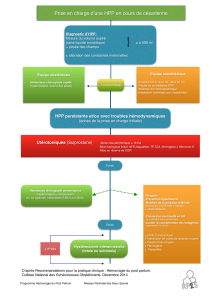
Néoplasie endocrinienne multiple de type 1 : une cause

OBSERVATION
Madame M., 57 ans, présentait une maladie lithiasique évoluant
depuis 1998 ayant nécessité 8 lithotripsies extra corporelles et 2
néphrolithotomies percutanées pour calculs rénaux bilatéraux et
récidivants.
Aucun bilan métabolique n’a été réalisé depuis le début de sa mal-
adie mais une analyse morpho-constitutionnelle a été réalisée sur
les calculs extraits lors de la dernière NLPC (décembre 2003). Cette
analyse était en faveur d’un calcul calcium-dépendant : Wheddelite
60%, Carbapatite 20% et Phosphate Octocalcique 10%. L’ensemble
faisant évoquer le diagnostic d’hyperparathyroïdie primaire (HPP).
L
’HPP était confirmée par le bilan biologique (calcium ionisé :
1.46mmol/l, PTH : 123 pg/ml, calcium total : 2.73 mmol/l,
phosphorémie : 0.9, calciurie des 24 heures : 390 mg). L’échogra-
phie parathyroïdienne et la scintigraphie au Sesta-MIBI objecti-
vaient deux nodules parathyroidiens supérieur et inférieur gauches
et un nodule froid thyroïdien droit.
La patiente était adressée en consultation d’endocrinologie compte
tenu de l’HPP et d’un syndrome dysmorphique clinique associé à une
hypertension artérielle, un diabète non insulino-dépendant, un syndro-
me du canal carpien et un syndrome d’apnée du sommeil. Une IRM
hypophysaire découvrait alors un processus intra-sellaire de 10 mm de
diamètre sans extension supra-sellaire. Biologiquement, l’IgF1 était à
389 ng/ml et l’hormone de croissance (STH ou GH) était élevée et
non-freinable par l’épreuve d’hyperglycémie provoquée par voie orale.
Le diagnostic d’Acromégalie était donc posé. L’ensemble des patholo-
gies endocriniennes s’inscrivait dans le cadre d’une Néoplasie Endo-
crinienne Multiple de type 1 (NEM 1) avec biologiquement une muta-
tion de la protéine A541 Tau niveau de l’Exon 10 du gène de la Méni-
ne. L’HTA avec discrète hypertrophie ventriculaire gauche, le syndro-
me d’apnée du sommeil et le syndrome du canal carpien correspon-
daient à des complications de l’acromégalie. Un scanner abdominal
n’a mis en évidence ni syndrome tumoral pancréatique ni syndrome
tumoral surrénalien.
La patiente bénéficiait en avril 2004 d’une parathyroïdectomie
supérieure et inférieure gauche avec thyroïdectomie totale et d’une
chirurgie trans-sphénoïdale en septembre 2004.
L’examen anatomo-pathologique confirmait les deux adénomes
parathyroïdiens à cellules principales et l’adénome somatotrope.
Aucun élément suspect de malignité n’a été noté sur la thyroïde
(multi-hétéro nodulaire avec remaniements fibreux et inflammatoi-
res).
En janvier 2005, la patiente présentait le bilan biologique suivant :
PTH sérique et bilan phospho-calcique normaux, IgF1 normalisé à
175 ng/ml et cycle GH normal freinable sous HGPO. L’IRM hypo-
physaire était sans particularité avec absence de reliquat tumoral
visible. Aucune récidivelithiasique n’a été observée.
LA NEOPLASIE ENDOCRINIENNE MULTIPLE DE TYPE 1
Les néoplasies endocriniennes multiples (NEM) se définissent,
chez un même sujet, par l’atteinte d’au moins deux glandes endo-
crines sans lien fonctionnel entre elles. Il en existe 3 types. La NEM
de type1 (ou syndrome de Wermer) est une association lésionnelle
héréditaire à transmission autosomique dominante. Les glandes
atteintes sont : les parathyroïdes, le pancréas endocrine, l’antéhy-
pophyse, la corticosurrénale et les tissus endocriniens situés dans
les bronches et le thymus. La majorité de ces tumeurs sont béni-
gnes.
Les tumeurs des parathyroïdes sont présentent dans 80 à 90% des
cas et sont habituellement responsables d’HPP [1-2].
◆
CAS CLINIQUE Progrès en Urologie (2006), 16, 376-377
Néoplasie endocrinienne multiple de type 1 : une cause possible de maladie
lithiasique rénale sévère et récidivante
Lamine NIANG (1),Mohamed Amine LACKMICHI (1),Sophie PERIE (2),Frédéric THIBAULT (1),
Bernard GATTEGNO (1),Philippe THIBAULT (1),Olivier TRAXER (1)
(1) Service d’Urologie, (2) Service d’ORL, Hôpital Tenon, Paris, France
RESUME
Les néoplasies endocriniennes multiples (NEM) se définissent, chez un même sujet, par l’atteinte d’au moins
deux glandes endocrines sans lien fonctionnel entre elles. Il en existe 3 types. La NEM de type 1 (ou syndrome de
Wermer) est une association lésionnelle héréditaire à transmission autosomique dominante.
Au travers d’une observation, nous rappelons les caractéristiques de la NEM de type 1 à l’origine d’une maladie
lithiasique sévère et récidivante et nous insistons sur la réalisation systématique d’un bilan étiologique pour tout
patient lithiasique dès le premier épisode selon les recommandations du Comité Lithiase de l’Association Fran-
çaise d’Urologie (CLAFU).
Mots clés : Lithiase, calcul, hyperparathyroïdie, acromégalie, calcium.
376
Manuscrit reçu : octobre 2005, accepté : décembre 2005
Adresse pour correspondance : Dr. O. Traxer, Service d’Urologie, 4, rue de la Chine,
Hôpital Tenon, 75020 Paris.
e-mail : olivier[email protected]
Ref : NIANG L., LACKMICHI M.A., PERIE S., THIBAULT F., GATTEGNO B., THI-
BAULT P., TRAXER O. Prog. Urol., 2006, 16,

La découverte d’une NEM chez un patient doit faire rechercher des
cas familiaux similaires. Ce ne fut pas le cas pour notre patiente.
Le gêne majeur de prédisposition de la NEM1 a été cloné en 1997,
il est constitué de 10 exons facilement accessible à une analyse
conformationnelle. Les exons sont entièrement séquencés. Une
mutation est retrouvée chez plus de 85% des patients présentant une
NEM1 cliniquement bien authentifiée, comme ce fut le cas pour
notre patiente [1-2].
DISCUSSION
Les manifestations cliniques des NEM dépendent du type de
tumeurs endocriniennes.
La lithiase urinaire est révélatrice d’une HPP dans environ 19% des
cas. Pour notre patiente le diagnostic d’HPP a été posé tardivement
(après sept récidives lithiasiques). Un bilan métabolique de premiè-
re intention selon les recommandations du CLAFU aurait pu faire
suspecter l’HPP [3]. Cette observation met l’accent sur l’importan-
ce de l’évaluation médicale de la maladie lithiasique. De la même
façon, l’analyse morpho-constitutionnelle des calculs par spectro-
photométrie infra-rouge doit être systématique. Dans cette observa-
tion, c’est l’analyse du calcul qui a fait suspecter l’HPP, mais là
encore cette analyse n’a été réalisée que 6 ans après le début de la
maladie lithiasique alors que la patiente avait déjà bénéficié d’une
NLPC. Enfin, la découverte d’une HPP doit systématiquement faire
évoquer la possibilité d’une NEM.
Pour notre patiente la découverte de l’acromégalie a été suggérée
devant le syndrome dysmorphique mais aussi l’HTA, le diabète et
le syndrome du canal carpien, complications habituelles de l’acro-
mégalie. L’élévation de l’IgF1 non freinée par l’HGPO confirme le
diagnostic d’acromégalie et l’IRM hypophysaire montre le plus
souvent un processus intracellaire comme pour notre patiente. Par-
fois, le processus est supra ou latéro-sellaire. L’adénomectomie
sélectivepar voie trans-phénoïdale reste le traitement de référence
de l’acromégalie. Pour notre patiente cette voie d’abord a permis
l’exérèse complète de l’adénome et la normalisation de l’IgF1 en
post-opératoire. La radiothérapie peut être indiquée en cas d’exérè-
se partielle de la tumeur et le traitement médical est préconisé pour
les jeunes patients dont les fonctions hypophysaires doivent être
respectées.
La prise en charge de ces patients doit donc être multidisciplinaire
avec participation des endocrinologues. Il est cependant essentiel de
rappeler que le bilan phospho-calcique initial peut et doit être réali-
sé par l’urologue qui reste souvent le premier et le seul interlocuteur
dans la maladie lithiasique. Enfin, il faut garder à l’esprit que le
retard diagnostique est source d’un surcoût dans la prise en charge
deces patients. Pour le cas présent, il est difficile de chiffrer ce sur-
coût mais il faut noter que cette patiente était prise en charge dans
notre service depuis de nombreuses années pour pathologie lithia-
sique sans qu’une évaluation métabolique n’ait été réalisée.
CONCLUSION
Notre observation met l’accent sur le bilan métabolique des patients
lithiasiques. Au travers d’une présentation clinique rare, nous sou-
haitons insister sur l’importance du bilan métabolique pour tout
patient lithiasique selon les recommandations du CLAFU. L’analy-
se morpho-constitutionnelle du calcul par spectro-photométrie
infra-rouge doit être systématique si le calcul est disponible. Une
prise ne charge multidisciplinaire des patients lithiasiques doit être
organisée pour une évaluation optimale de la maladie lithiasique.
Enfin, la découverte d’une hyperparathyroïdie doit systématique-
ment faire rechercher une NEM en vue d’un diagnostic et d’une
prise en charge précoces.
REFERENCES
1. THE EUROPEAN CONSORTIUM ON MEN1. Identification of the multi-
ple endocrine neoplasia type1: European consortium on MEN1. Hum. Mol.
Genet., 1997 ; 6 : 1177-1183.
2. CALENDER A., GOUDET P. et le Groupe d'Étude des Néoplasies
EndocriNIENNES MULTIPLES (GENEM). Les néoplasies endocriniennes
multiples de type 1 : NEM1-Syndrome de Wermer. Ann. Endocrinol., 2003;
64 : 383-388.
3. TRAXER O.:Le bilan des patients lithiasiques : Quand,comment et pour-
quoi ? Prog Urol FMC 2000 ; 10 : 3-10.
____________________
SUMMARY
Multiple endocrine neoplasia type 1: a possible cause of severe and
recurrent urolithiasis.
Multiple endocrine neoplasia (MEN) is defined by the presence of at
least twofunctionally unrelated endocrine gland tumours in the same
subject. There are three types of MEN. MEN type 1 (or Wermer syndro-
me) is an autosomal dominant cancer syndrome. In the light of a case
report, the authors recall the characteristics of MEN type 1 responsible
for severe and recurrent urolithiasis and emphasize the need for syste-
matic aetiological work-up for all patients presenting a first episode of
urolithiasis according to the guidelines of the Association Française
d’Urologie Stone Committee (CLAFU).
Key words: urolithiasis, calculi, hyperparathyroidism, acromegaly, cal-
cium.
L. Niang et coll., Progrès en Urologie (2006), 16, 376-377
377
____________________
1
/
2
100%