Partie 1 - PlanetHoster

Lycée Saint Martin
5 Cloître Saint Martin - BP 32209
49022 Angers cedex 02
Tel:02 41 88 09 00
SVT
DEVOIR SURVEILLÉ
NOM :
Prénom :
Classe : 1ere S
:: Stabilité et variation du patrimoine génétique ::
A lire attentivement avant de commencer !
Vous porterez une attention particulière à l'organisation des réponses sur votre copie
et à la présentation de ses réponses.
Rappelez-vous les indications données lors de la correction du dernier devoir.
Si les critères de lisibilité qui ont été indiqués ne sont pas respectés,
votre note finale ne pourra en aucun cas dépasser la moyenne.
Rédigez sur copie double.
Rendre la page 2 du sujet.
Cochez cette case pour indiquer que vous avez lu et compris l'avertissement ci-dessus.
Partie 1 – Restitution organisée des connaissances [10 points] (prévoir 40 à 45 minutes)
« A l'issue d'un cycle cellulaire, les deux cellules-filles possèdent un programme
génétique identique à celui de la cellule-mère »
Discutez cette affirmation : montrez qu'elle est juste d'une certaine façon,
mais qu'elle peut être fausse d'une autre façon.
•Construisez votre argumentation sous la forme d'un texte comportant deux
parties distinctes. Séparez chacune de ces parties.
•Chaque partie ne traitera que d'un seul élément de discussion. Titrez chacune de
ces parties. Le titre fera référence à l'idée développée dans la partie.
•Les mots et expressions suivantes devront apparaître dans votre écrit et mis en
évidence par soulignage. Au moins quatre autres mots ou expressions « clés »
seront également soulignées.
Anaphase - chromatide - caryotype – interphase - réplication – appariement des
nucléotides
•Attention ! Il ne s'agit pas de « réciter son cours » mais plutôt de rédiger un écrit
personnel à partir des connaissances acquises et qui apporte une réponse au sujet
figurant dans l'encadré ci-dessus.
Vous pouvez utiliser le verso de cette page comme brouillon
© JMH 21/11/2013 - http://ent.sapiens-jmh.planethoster.org 01S _2013-11-20.odt page 1 / 3

Feuille à rendre avec votre copie
Nom : .............................................................. Prénom : .......................................................
Partie 2 – Exploitation de documents [10 points] (prévoir 10 à 15 minutes)
Une personne s'intéresse à un phénotype pathologique : la phénylcétonurie, maladie dont est
atteinte l'un de ses amis. Les informations qu'il a trouvées sont données dans les documents qui
suivent.
A partir de ces informations que vous mettrez en relation avec vos connaissances, vous cocherez pour
chacun des items ci-dessous, la proposition qui vous paraît correcte.
Item 1 – A l'échelle
macroscopique le phénotype
phénylcétonurique est caractérisé
par :
un foie anormalement gros
un cerveau de masse faible
un volume de sang très important
aucun caractère observable
Item 2 – A l'échelle
microscopique, le phénotype
phénylcétonurique est caractérisé
par :
des cellules sanguines anormales
des cellules du foie anomales
des neurones anormaux
Aucun caractère observable
Item 3 – A l'échelle moléculaire,
le phénotype phénylcétonurique
est caractérisé par :
un excès de phénylalanine
une insuffisance de myéline
une PAH peu ou pas active
un ADN anormal
Item 4 – La phénylcétonurie :
n'est pas d'origine génétique, mais d'origine alimentaire
est d'origine génétique. Elle provient d'une mutation
C315T
est d'origine génétique. Elle provient d'une mutation de
type insertion des nucléotides ATC en position 283
est d'origine génétique. Elle provient d'une mutation de
type délétion des nucléotides ATC en position 283
Item 5 – La phénylcétonurie est une maladie :
non héréditaire
héréditaire qui implique plusieurs gènes (PAHn, PAHm1,
PAHm2)
qui touche les personnes homozygotes pour l'allèle
PAHm1
qui touche les personnes hétérozygotes pour le gène de
la PAH
Document A – Les symptômes cliniques de la maladie sont une grave arriération psychique et des
troubles caractériels. La plupart des sujets atteints de phénylcétonurie et non traités, sont victimes d'un
retard mental extrêmement sévère. La masse du cerveau de ces individus est au-dessous de la normale.
Une cellule nerveuse (ou neurone) normale possèdent des prolongements membranaires et
cytoplasmiques appelés fibres nerveuses. Chez certains neurones, l'un de ces prolongements appelé
axone, est entouré d'une gaine de myéline qui joue un rôle dans la propagation des messages nerveux.
Chez un patient atteint de phénylcétonurie, la myélinisation (=formation de la gaine de myéline) de leurs
nerfs est défectueuse et leurs réflexes sont hyperactifs.
La phénylcétonurie est une maladie qui touche environ un nouveau-né sur 16 000, en France.
L'espérance de vie des phénylcétonuriques non traités est considérablement raccourcie. La moitié d'entre
eux sont morts à l'âge de vingt ans, et les trois quarts à l'âge de trente ans.
Cette atteinte du cerveau est due à l'effet toxique de la phénylalanine, un acide aminé, présent en excès
dans le plasma sanguin. Chez certains malades ce taux peut atteindre 1 000 µMole.L-1 de plasma, alors
qu'il devrait être normalement de l'ordre de 60 µMole.L-1 de plasma.
La phénylalanine est apporté par l'alimentation est est normalement transformé en tyrosine (un autre
acide aminé) dans les cellules du foie. Cette transformation est catalysée par une enzyme, la PAH
(PhénylAlanineHydroxylase). La concentration plasmatique en phénylalanine dépend du taux d'activité de
l'enzyme : elle est élevée lorsque l'activité de la PAH est inférieure à 1%. Au-dessus de 5%, elle est
modérée.
Un test simple, réalisé à partir de quelques gouttes de sang, permet de savoir si un enfant présente ou
non de cette maladie. Si la maladie est dépistée de façon précoce, il est possible d'en minimiser les effets
en adoptant un régime alimentaire pauvre en phénylalanine (fruits et légumes essentiellement).
© JMH 21/11/2013 - http://ent.sapiens-jmh.planethoster.org 01S _2013-11-20.odt page 2 / 3

Document B – Le gène PAH présent dans le génome d'un être humain est porteur d'une information
à partir de laquelle les cellules du foie produisent une enzyme appelée PAH. Une étude épidémiologique a
montré la présence de l'allèle PAHn chez les personnes non atteintes de phénylcétonurie et de l'allèle
PAHm1 ou de l'allèle PAHm2 chez les personnes atteintes.
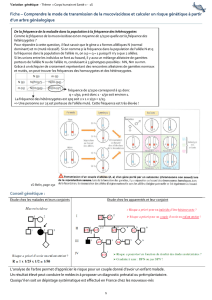
Document C – Arbre généalogique et analyse génétique des membres d'une famille
All = Allèle
© JMH 21/11/2013 - http://ent.sapiens-jmh.planethoster.org 01S _2013-11-20.odt page 3 / 3
1
/
3
100%