Télécharger le document au format PDF

Le système endocrinien de l’homme adulte trouve son unité
dans le vecteur de ses messages :l’hormone [87]. Un androgè-
ne est une hormone stéroïdienne capable d’induire la différen-
tiation et la maturation des organes reproducteurs masculins, de
stimuler les caractères sexuels secondaires pour aboutir à un
phénotype masculin normal [114] et d’entraîner les modifica-
tions comportementales nécessaires au rôle de l’homme dans la
reproduction [23]. La testostérone, hormone mâle sécrétée par le
testicule, est l’androgène majeur qui exerce une action quasi-
ubiquitaire dans l’organisme de l’homme, directement ou par
l’intermédiaire de sa bioconversion en un androgène plus puis-
sant, la dihydrotestostérone (DHT) ou en un œstrogène puissant,
l’œstradiol (E2). Le maintien d’un niveau approprié d’androgè-
nes impose une production qui équilibre l’épuration métabo-
lique et l’excrétion.
I. LA TESTOSTERONE : SYNTHESE,
REGULATION, TRANSPORT ET
METABOLISME
1- Biosynthèse des androgènes
a) Testostérone
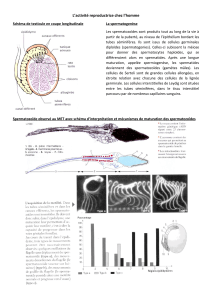
La testostérone est le principal androgène circulant. Elle est pro-
duite de façon quasi-exclusive (plus de 95%) par les cellules de
Leydig du testicule, situées autour et entre les tubes séminifères,
qui représentent 5% du volume de la glande [153]. Le nombre
maximum de cellules de Leydig, atteint peu après 20 ans, est de
500 millions par testicule [121]. La sécrétion globale de testos-
térone est de 5 à 7,5 mg/24h chez l’homme adulte normal [185].
Des androgènes sont également synthétisés en faibles quantités
par la surrénale et en quantités infinitésimales par le cerveau
[10, 83] où l’action locale pourrait cependant être importante.
Le contenu en testostérone du testicule de l’homme adulte est
d’environ 1µg/g de testicule, ce qui montre que la quasi-totalité
de la testostérone secrétée est libérée dans la circulation [193].
Le précurseur des androgènes est le cholestérol. Les cellules de
Leydig peuvent en assurer la synthèse à partir de l’acétate, mais
elles utilisent surtout le cholestérol extrait des lipoprotéines
plasmatiques, et notamment de la fraction de faible densité
(LDL) [140], mais aussi celui des membranes cellulaires (Figu-
re 1). Le cholestérol (C27) est transporté vers les mitochondries
par un mécanisme dépendant de la LH régulé par une protéine
de transfert dite protéine activatrice de la stéroïdogénèse (StAR
ou steroidogenesis activator protein) [169] dont le rôle essentiel
aégalement été décrit dans la surrénale et l’ovaire [170] ainsi
que le cerveau [83]. Ce transfert intra-mitochondrial du choles-
térol est l’étape limitante de la stéroïdogenèse. PBR (peripheral
benzodiazepine receptor) y participe de façon minoritaire [96].
L’absence de StAR par mutation du gêne est responsable de
l’hyperplasie surrénale congénitale lipoïde, caractérisée par une
accumulation de cholestérol dans les cellules de Leydig et de la
surrénale et une quasi-impossibilité de synthétiser des stéroïdes
[168]. Dans la mitochondrie, le début de la cascade de la stéroï-
dogenèse est marqué par le clivage du cholestérol (C27) en pré-
gnénolone (C21) par le cytochrome P450scc (side-chain cliva-
ge). La prégnénolone, biologiquement inactive, est éjectée dans
le réticulum endoplasmique où elle est métabolisée, notamment
sous l’action d’enzymes oxydatifs du groupe des cytochromes
P450. La prégnénolone est alors convertie en une variété de sté-
roïdes C19. Deux voies sont possibles avant d’aboutir à la tes-
tostérone, désignées sous les termes de voie ∆4 ou ∆5, suivant
que les composés intermédiaires sont des 3-céto, ∆4stéroïdes ou
des 3-hydroxy, ∆5stéroïdes (Figure 2) :
-la voie préférentielle dans le testicule est la voie ∆5qui fait
intervenir en premier P450C17 qui est codé par le gène
CYP17 [213] dont l’expression est sous le contrôle de la LH.
Le premier composé intermédiaire est la 17α-hydroxypré-
Progrès en Urologie (2004), 14, 639-660
Physiologie des androgènes chez l’homme adulte
J. TOSTAIN, D. ROSSI, P.M MARTIN1
1Laboratoire de transfert en oncologie biologique, CHU de Marseille
639
◆Physiologie des Androgènes :
de l’Homme Adulte à l’Homme Vieillissant
Et que derrière un voile, invisible et présente,
J’étais de ce grand corps l’âme toute puissante.
Jean Racine (Britannicus)

gnénolone. Le clivage de la chaîne latérale fait apparaître le
premier stéroïde C19, la déhydroépiandrostérone ou DHEA.
L’action du complexe 3β-hydroxystéroide déshydrogéna-
se/∆5-∆4-isomérase (3βHSD) la transforme en ∆4-androstè-
nedione, puis celle de la 17-β-hydroxystéroide déshydrogé-
nase (17βHSD réduisant le groupe cétonique 17 en OH)
aboutit à la formation de testostérone.
-la voie accessoire est la voie ∆4qui fait intervenir en premier
le complexe 3β-hydroxystéroïde déshydrogénase ∆5-∆4-iso-
mérase pour former la progestérone qui est hydroxylée en
17α-hydroxyprogestérone. L’action de clivage de P450C17 la
transformera en ∆4-androstènedione, premier C19 de cette
voie minoritaire.
La LH intervient donc dans la régulation de la stéroïdogenèse à
deux niveaux :
-transfert du cholestérol de la membrane externe vers la mem-
brane interne de la mitochondrie faisant intervenir StAR dans
un processus de régulation à court terme
-activité des systèmes enzymatiques du réticulum endoplas-
mique assurant la transformation de la prégnénolone pour
une régulation à long terme.
La capacité du système enzymatique ne lui permettant pas de
transformer la totalité de la prégnénolone en testostérone, la cel-
lule de Leydig élimine des composés intermédiaires : DHEA,
progestérone, 17α-hydroxyprogestérone et androstènedione
[176].
b) DHEA et Sulfate de DHEA (SDHEA)
La DHEA et l’androstènedione sont également produits par la
surrénale. Ce sont des androgènes dits faibles en raison de la
modestie de leur action androgène et/ou de leur conversion pos-
sible en androgènes forts. Par exemple, moins de 1% de la tes-
tostérone sérique provient de la DHEA. Cependant, à l’intérieur
même des cellules (intracrinologie), 30 à 50% de la synthèse des
androgènes proviendraient de la transformation de la DHEA
[89], avec une formation pratiquement ubiquitaire de stéroïdes
sexuels [51, 77, 107], notamment dans le foie, la peau, la pro-
state, l’os et le cerveau qui possèdent le matériel enzymatique
nécessaire à cette transformation. La DHEA est en outre un
neurostéroïde associé à des interactions avec les neurotransmet-
640
Figure 1 : Régulation de la stéroïdogenèse dans la cellule de Leydig

641
Figure 2 : Biosynthèse des stéroïdes sexuels chez l’homme. Les flèches rouges pleines indiquent la voie préférentielle de la stéroïdogenèse
dans les cellules de Leydig (Abréviations : cyt P450scc : enzyme de clivage de la chaîne latérale du cholestérol ; 3
β
β
HSD : 3
β
β
-hydroxystéroi-
de déshydrogénase/
∆
∆
5-
∆
∆
4isomérase). La flèche bleue discontinue indique les réactions catalysées par la 3
β
β
HSD et sépare schématiquement
la voie
∆
∆
5 à gauche de la voie
∆
∆
4 à droite (17
β
β
HSD : 17
β
β
-Hydroxystéroïde déshydrogénase). La flèche rouge discontinue indique les réac-
tions catalysées par cette enzyme.

teurs cérébraux [9, 90, 179]. La présence d’un possible récep-
teur périphérique membranaire dans les cellules endothéliales
[97] et dans les cellules musculaires lisses de la paroi artérielle
[188] a été récemment suggérée.
Le SDHEA n’a pas de rôle spécifique mais, en en raison de sa
demi-vie longue (7-8h contre 15-30 minutes pour la DHEA) et
de son interconversion continuelle avec la DHEA [5], elle cons-
titue une réserve importante de DHEA.
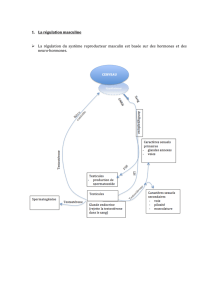
2- Régulation des androgènes
La quantité d’androgènes du sérum utilisable par les tissus est
principalement régulée par l’axe hypothalamo-hypophysaire par
l’intermédiaire de l’action de la LH sur les cellules de Leydig.
Durant la période fœtale vers la 14e semaine, on observe une
augmentation très importante du nombre et de l’activité des cel-
lules de Leydig sous l’influence de l’hCG maternelle [118].
Avant cette période, le contrôle des cellules de Leydig et le
début de la stéroïdogenèse seraient sous la dépendance d’un
autre mécanisme [140]. Dans la période néo-natale, l’action des
gonadotrophines amène les taux de testostérone au niveau de
ceux observés durant la puberté, puis les cellules de Leydig
régressent. Durant la puberté, l’augmentation des taux de LH
entraîne une deuxième phase de prolifération et de différentia-
tion conduisant au nombre de cellules de Leydig de l’adulte
[148]. La diminution de la testostérone liée à l’âge est pour par-
tie liée à l’atrophie progressive et à la disparition des cellules de
Leydig mais également au déclin des capacités de stéroïdogenè-
se [125].
La fonction principale des cellules de Leydig est la production
d’androgènes, mais elles produisent également de l’IGF-1 et des
facteurs de croissance qui interviennent dans la régulation auto-
crine et paracrine du testicule [141].
a) L’axe hypothalamo-hypophysaire
L’hypothalamus siège à la base du cerveau juste au dessus de
l’hypophyse et possède des liens très fournis avec les autres
régions cérébrales utiles aux fonctions viscérales, autonomes et
comportementales. L’éminence médiane et l’infundibulum,
richement vascularisées, assurent la communication neurolo-
gique et humorale entre l’hypothalamus et l’hypophyse (Figure
3). La partie postérieure de l’hypophyse (posthypophyse ou
neurohypophyse) joue le rôle de réservoir pour la vasopressine
et l’oxytocine qui sont sécrétées par l’hypothalamus. La partie
antérieure de la glande pituitaire (antéhypophyse ou adénohy-
pophyse) sécrète plusieurs hormones : les gonadotrophines,
FSH (follicle-stimulating hormone ou hormone folliculo-stimu-
lante) et LH (luteinizing hormone ou hormone lutéinisante),
l’ACTH, la prolactine, la GH et la TSH.
1. LES GONADOTROPHINES
Les cellules gonadotropes antéhypophysaires sécrétent les gona-
dotrophines LH et FSH. Ce sont des glycoprotéines qui inter-
viennent dans la régulation de la production d’androgènes. La
majorité des cellules gonadotropes peuvent sécréter les deux
gonadotrophines qui contiennent toutes les deux une chaîne αet
une chaîne β. Les chaînes αsont composées de 96 acides ami-
nés et sont identiques pour FSH, LH, TSH et hCG. La chaîne β
est par contre spécifique à chacune de ces hormones. Schémati-
quement, la FSH favorise la spermatogenèse alors que la LH sti-
mule la sécrétion d’androgènes par les cellules de Leydig.
2. RÉGULATION DE LA SÉCRÉTION DE LH
Elle fait intervenir une boucle de contrôle complexe équilibrant
les actions stimulantes des sécrétions hypothalamiques et l’ac-
tion frénatrice des stéroïdes sexuels (Figure 4).
• STIMULATION PAR LES HORMONES HYPOTHALAMIQUES
La LHRH (luteinizing hormone-releasing hormone) ou GnRH
(gonadotropin-releasing hormone ou gonadoréline) est sécrétée
par l’hypothalamus et stimule les cellules gonadotropes hypo-
physaires par l’intermédiaire d’un récepteur (GnRH-R) [155]
pour sécréter de la LH et, à un moindre degré, de la FSH. La
LHRH est un décapeptide libéré par l’hypothalamus sur un
mode pulsatile, donnant un caractère pulsatile et rythmique à la
sécrétion de gonadotrophines hypophysaires [120]. La baisse de
la testostérone après castration ou lors de l’insuffisance leydi-
gienne primitive, stimule la sécrétion de GnRH, principalement
en augmentant la fréquence des pulses [27]. A l’inverse, une sti-
mulation continue par la LHRH ou un analogue (du type de ceux
utilisés pour le traitement palliatif du cancer de prostate) entraî-
ne une « désensibilisation » des récepteurs avec une suppression
des ARNm de la LH-βet de la FSH-β[37].
Dans le sang périphérique, l’aspect pulsatile de la libération de
LH est plus clairement évident que pour la FSH dont la 1/2 vie
est plus longue. Les pulses sont irrégulièrement espacés et
d’amplitude variable (Figure 5). L’existence d’un rythme circa-
dien marqué par une élévation de l’amplitude des pulses de LH
642
Figure 3 : Axe neuro-endocrine hypothalamo-hypophysaire. Les cel-
lules hypothalamiques neurosécrétoires libèrent la LHRH qui par-
vient aux cellules gonadotropes par le réseau capillaire portal long.
Les cellules gonadotropes sécrètent et libèrent en réaction la LH
dans le réseau veineux

durant le sommeil et une augmentation de la testostérone aux
premières heures du matin, est bien établi chez l’adolescent
[19]. Par contre chez l’adulte on observe bien un rythme simi-
laire pour la testostérone, mais il est beaucoup moins évident
pour la LH [173], ce qui semblerait indiquer l’existence d’un
autre mécanisme régulateur. Le rythme diurne de la testostérone
est perturbé par la fragmentation du sommeil [103] et s’émous-
se lors du vieillissement [20]. L’influence de ces rythmes diurne
et pulsatile sur la régulation des protéines est inconnue.
2. INHIBITION PAR LES STÉROÏDES SEXUELS
L’action frénatrice (feedback négatif) des stéroïdes sexuels sur
la libération des gonadotrophines s’exerce au niveau hypothala-
mique et hypophysaire. La LH et la FSH plasmatiques s’abais-
sent en quelques heures [151] après administration de testosté-
rone ou d’oestradiol alors que la castration les élèvent. La tes-
tostérone diminue l’expression de la sous-unité du gène de la
LH [195], mais l’inhibition la plus importante est exercée par la
testostérone, la DHT et l’oestradiol au niveau de l’hypothalamus
en ralentissant le générateur de pulses hypothalamique et donc
la libération de LH [151, 197]. La castration chirurgicale, faisant
disparaître le rétro-contrôle négatif, entraîne une augmentation
marquée de FSH et de LH.
Il est possible qu’une grande partie de l’action inhibitrice de la
testostérone s’exerce au travers de sa bioconversion en oestra-
diol [151, 158], d’autant que l’oestradiol réduit également l’am-
plitude des pulses de LH chez l’homme [151] et a un effet inhi-
biteur direct sur les cellules gonadotropes hypophysaires [81].
La testostérone inhibe préférentiellement la sécrétion de LH, alors
que les oestrogènes, dont l’action frénatrice globale est plus mar-
quée que celle de la testostérone [39], inhibent de façon identique
la LH et la FSH. L’inhibine, une protéine produite par les cellules
de Sertoli, est aussi capable d’inhiber la production de FSH, mais
n’a que peu ou pas d’effet sur la sécrétion de LH [24].
3. AUTRES FACTEURS DE RÉGULATION
Le système nerveux central pourrait réguler la sensibilité des
cellules de Leydig à la LH [42, 154]. Chez le rat, les hormones
thyroïdiennes stimulent l’expression de StAR et la production
de stéroïdes [106], alors que les glucocorticoïdes ont plutôt une
action inhibitrice de la stéroïdogenèse en induisant l’apoptose
des cellules de Leydig, ce qui pourrait contribuer à l’abaisse-
ment des androgènes lors du stress [57].
L’effet de la prolactine sur les cellules de Leydig est débattu
[105]. L’abaissement de la testostérone observée chez les hom-
mes présentant un adénome hypophysaire à prolactine est prin-
cipalement dû à la suppression de la sécrétion pulsatile de LH
[198].
L’ACTH stimule la sécrétion d’androgènes surrénaliens faibles.
En retour, ceux-ci n’exercent aucun rétrocontrôle sur la sécré-
tion d’ACTH qui est régulée par le cortisol.
b) Mode d’action de la LH
La LH agit sur les cellules de Leydig par l’intermédiaire de
récepteurs (LH-R) qui font partie de la superfamille des récep-
teurs couplés à la protéine G (GPCR) [109]. Le ligand naturel du
récepteur à la LH est la LH, mais en raison d’analogies structu-
relles, la gonadotrophine chorionique humaine (hCG) peut éga-
lement l’activer. En pathologie clinique, des mutations de ces
récepteurs affectent la production d’androgènes : activation
responsable d’une puberté précoce ou, à l’inverse, inactivation
entraînant un pseudo-hermaphrodisme masculin.
L’activation des récepteurs de la LH au niveau de la membrane
des cellules de Leydig stimule l’adényl-cyclase et entraîne la
formation d’AMP cyclique (cAMP). Le cAMP active les protéi-
nes kinases qui favorisent la conversion du cholestérol en pré-
gnénolone. Il est possible que le calcium et la calmoduline puis-
sent aussi intervenir comme second messager de l’action de la
LH [141] (Figure 1).
A côté cette action rapide sur la stéroïdogenèse, la LH possède
643
Figure 4 : Boucles de contrôle de l’axe hypothalamo-hypophyso-tes-
ticulaire
Figure 5 : Taux de LH (cercles pleins) et de sa sous-unité
α
α
(carrés
vides) dans le sang périphérique d’un homme adulte sain (Prélève-
ment toutes les 10 minutes entre 8h00 et 20h00). D’après Winters
[194]
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
1
/
50
100%