La mucoviscidose

http://perso.ens-lyon.fr/romain.berardozzi/
LA MUCOVISCIDOSE : ASPECTS CLINIQUES, CELLULAIRES,
MOLECULAIRES ET GENETIQUES
La mucoviscidose est une pathologie fréquente en France et Amérique du Nord (rare en Afrique et
Asie) affectant près d’une personne sur 2500. La morbidité est élevée en l’absence de traitement et
l’espérance de vie moyenne d’un patient atteint de mucoviscidose est de 47 ans (contre 7 ans en
1965). L’expressivité de la pathologie est très variable (chiffres Institut Pasteur).
Quels sont les phénotypes macroscopique, cellulaire et moléculaire des patients atteints de
mucoviscidose ?
Quelles sont les causes de cette pathologie ? En quoi cette pathologie diffère-t-elle d’autres pathologies
comme le cancer ou le diabète ?
Quelles sont les stratégies thérapeutiques actuelles ?
La mucoviscidose, une pathologie affectant les éptihélia glandulaires (voies
respiratoires, digestives, reproductrices et sudoripares)
Tableau clinique d’un patient atteint de mucoviscidose
Description du phénotype macroscopique (Tableau 1). De nombreuses fonctions biologiques sont
déficientes chez un patient atteint de mucoviscidose.
Comment expliquer une telle généralisation des symptômes ?
Un mucus visqueux à l’origine des symptômes de la mucoviscidose ou fibrose kystique
L’étude par radiographie aux rayons X (Figure 1) et par préparation de lame histologique de bronches
de patients atteints de mucoviscidose (Figure 2) montre la présence de mucus abondant et visqueux
à la surface de l’épithélium bronchique qui obstrue la bronche.
De manière générale, tous les épithélia glandulaires exocrines (glande exocrine du pancréas
(acini), foie exocrine, vésicule biliaire, intestin, glandes sudoripares, canaux déférents) sont affectés et
produisent un mucus visqueux dont les propriétés rhéologiques sont en défaveur de sa clairance.
Cela aboutit à une accumulation de mucus et à un tapissage des épithélia (réduisant leur capacité
d’échange : malabsorption intestinale, difficultés respiratoires Tableau 1) voire une obstruction de la
lumière des épithelia et des canaux associés (obstruction bronchique, atrésie des canaux déférents,
calculs biliaires Figure 4, Tableau 1). L’obstruction aboutit à une perte de fonctionnement de l’organe
et à sa dégradation (cirrhose hépatique, fibrose pancréatique et kystes pancréatiques (fibrose
kystique) par action des enzymes digestives non libérées dans le duodénum Figure 4, atteinte
secondaire du pancréas endocrine aboutissant à des diabètes).
La réduction de la clairance muco-ciliaire des voies respiratoires supérieures entraine la colonisation
bactérienne du mucus expliquant les infections respiratoires à répétition (Pseudomonas aeruginosa,
Staphylococcus aureus, Haemophilus influenzae).
Quelles sont les bases cellulaires et moléculaires permettant d’expliquer la formation d’un mucus
visqueux ?

http://perso.ens-lyon.fr/romain.berardozzi/
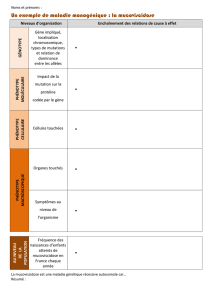
Tableau 1 : Tableau clinique d’un patient atteint de mucoviscidose : phénotype à l’échelle
macroscopique. De nombreuses fonctions biologiques sont détériorées chez le patient malade. Les
organes affectés sont tous des épithélia glandulaires. Les conséquences physiologiques sur les
organes concernés sont à mettre en relation avec la viscosité du mucus qui recouvre voire bouche les
canaux ménagés par les épithélia.
Figure 1 : Clichés de radiographies aux rayons X d’un patient sain (gauche) et
d’un patient atteint de mucoviscidose (droite). Remarquer à droite chez le
patient atteint de mucoviscidose la présence au niveau des bronches de matériel
opaque aux rayons X (blanc sur le cliché comme les os).
Figure 2 : Coupe
transversale de bronche
mettant en évidence les
caractéristiques des
epithélia bronchiques chez
un patient atteitnde
mucoviscidose (A, haut) et
un patient sain (B,bas).
Remarquer chez le patient
atteint de mucoviscidose la
présence dans la lumière des
bronches de mucus. Ce
mucus abondant n’est pas
évacué par clairance muco-
ciliaire. C’est ce mucus qui est
observé en Figure 1 et qui est
opaque aux rayons X.
Figure 3 : Coupe
transversale de l’iléon d’un
porc sain (gauche) et d’un
porc atteint de
mucoviscidose (droite)..
L’iléon est la partie terminale
de l’intestin grêle juste en
amont du colon. On remarque
chez le porc atteint de
mucoviscidose une obstruction
de la lumière intestinale par un
bouchon de mucus. Cette
obstruction explique les
symptômes digestives et
notamment la malabsorption
des nutriments conduisant à
une malnutrition voire
dénutrition.
Figure 4: Pancréas et vésicule biliaire chez un porc sain (clichés de gauche sur les
deux images) et un porc atteint de mucoviscidose (clichés de droite). On remarque une
dégradation du pancréas résultat de l’accumulation intra-tissulaire des enzymes pancréatiques
non libérées qui attaquent les cellules exocrines et endocrines (diabètes). La vésicule biliaire
chez le porc malade est nécrosée et ne permet plus le stockage de la bile nécessaire à
l’émulsion des lipides et par la suite leur dégradation par les lipases. Ces observations mettent
en évidence l’existence de troubles digestifs chez les patients atteints de mucoviscidose.
Figure 5:Arbre généalogique d’une famille porteuse de la mutation CFTR
responsable de la mucoviscidose. Remarquer le caractère autosomique récessif
de la transmission. A partir de ce mode de transmission, on en déduit qu’un couple
de porteurs sains à une probabilité d’avoir un enfant malade de ¼ .
Le génotype des parents porteurs sains est CFTR-/CFTR+ (avec CFTR moins le
gène muté).
Le génotype des malades est CFTR-/CFTR-.
Le génotype des individus sains est CFTR+/CFTR+.

http://perso.ens-lyon.fr/romain.berardozzi/
La mucoviscidose, une pathologie héréditaire liée à la mutation du gène codant pour
la protéine CFTR
Une pathologie génétique transmise de génération en génération
L’étude de nombreux arbres généalogiques de familles dont un parent est atteint de mucoviscidose
(Figure 5) permet d’observer que :
1. la probabilité d’être atteint de la mucoviscidose est plus forte lorsqu’un parent au moins est atteint
la mucoviscidose est une maladie héréditaire (pathologie conséquence d’une anomalie génétique
transmise de génération en génération)
2. les hommes (XY) et les femmes (XX) sont indifféremment affectés par la pathologie la mutation
responsable de la pathologie n’est pas portée par le chromosome X (sinon 100% mâles (XY ; un
unique X) serait atteint de la maladie): la pathologie est transmise par voie autosomique, le gène muté
est porté par un autosome.
3. des parents sains donnent naissance à un enfant malade avec une probabilité de ¼ la mutation
d’une copie du gène sur les deux présentes n’entraine pas l’apparition d’un phénotype « malade ». Il
existe des porteurs sains : la mutation responsable de la mucoviscidose est récessive.
Quel est le gène muté ? Où est-il positionné dans le génome ?
Une pathologie liée à la mutation d’un gène porté par le chromosome 7
Les méthodes de génétique inverse ont permis de positionner le locus du gène muté chez les patients
atteints de mucoviscidose (Figure 6)
Pour quelle protéine code le gène muté chez les patients atteints de mucoviscidose et quelle est sa
fonction ? Comment la mutation du gène altère la fonction de la protéine ?
Une pathologie liée à un défaut de la glycoprotéine canal à anions CFTR
Les études de génétique inverse ont permis de mettre en évidence que la protéine mutée chez les
patients atteints de mucoviscidose est la protéine CFTR (Cystic Fibrosis Transmembrane
conductance Regulator) (Figure 7). Cette protéine est une glycoprotéine transmembranaire : un
canal à anions de la membrane plasmique des cellules épithéliales. Son ouverture est dépendante
de la fixation d’ATP (pas d’hydrolyse) sur le domaine NBD et est déclenchée par la phosphorylation
par la protéine kinase A du domaine de régulation.
Cette protéine permet la sortie d’ions chlorure en dehors de la cellule. La sortie d’ions chlorure dans
la lumière des épithélia glandulaires entraine par phénomène d’osmose la sortie d’eau ce qui fluidifie
le mucus et/ou les sécrétions enzymatiques. Ce phénomène est appelé transport hydro-électrique.
La sortie d’eau est amplifiée grâce à l’inhibition des canaux sodiques par CFTR. Le sodium ne rentre
alors plus dans la cellule ce qui augmente la pression osmotique extracellulaire et augmente la sortie
d’eau (Figure 8).
Le canal CFTR permet également la sortie d’ions thiocyanates. Ces ions forment dans la lumière des
bronches de l’hypothiocyanate ayant un rôle antimicrobien.
On distingue différentes mutations chez les patients atteints de mucoviscidose (Figure 9):
+mutation non-sens (codon stop précoce) ou défaut d’épissage conduisant à une protéine tronquée
puis rapidement dégradée
+mutation empêchant le repliement correct, la maturation et l’adressage de la protéine à la membrane
plasmique conduisant à une accumulation dans le Golgi et les vésicules d’exocytose (délétion de la
phénylalanine 508) : cette mutation est la plus fréquente, 87% des patients atteints de
mucoviscidose. (Figure10, Figure 11)
+mutation du domaine R bloquant les phénomènes de régulation de la protéine
+mutation empêchant le passage des ions (trouble de la conductance)

http://perso.ens-lyon.fr/romain.berardozzi/
Figure 9:Différentes mutations à l’origine de la mucoviscidose.
La formation d’un mucus visqueux a pour origine une défaillance de la sortie des ions chlorures
dans la lumière des épithélia. L’absence de sortie des ions chlorure peut s’expliquer l’absence de
la protéine, son mauvais repliement/adressage à la membrane plasmique (voir Figure 10), à des
défauts dans le domaine R de régulation ou encore à des défauts de conductance.
Figure 10:Localisation par immunofluorescence de la protéine CFTR chez un patient sain
(gauche) et un patient atteint de mucoviscidose porteur de la délétion de la Phe508 (droite).
L’utilisation d’anticorps dirigés contre la protéine CFTR et couplé à des molécules fluorescentes
permet grâce à l’observation par microscopie de fluorescence de localiser la protéine au sein des
cellules de l’épithélium bronchiques.
On observe chez le patient sain que le protéine CFTR est localisée à la membrane apicale des
cellules de l’épithélium. Chez le patient malade porteur de la délétion Phe508 (à droite), on
observe que la protéine est localisée dans l’ensemble de la cellule (plus particulièrement dans le
RER, le Golgi et les vésicules d’endocytose). En effet, cette délétion a pour conséquence un
défaut de repliement de la protéine qui reste alors dans le RER et d’adressage à la membrane
plasmique et qui reste alors dans le Golgi ou les vésicules d’exocytose.
Figure 7:Structure de la protéine CFTR.
La protéine CFTR est une protéine canal transmembranaire composée de 12 hélices alpha hydrophobes. Ces hélices
aménagent un espace hydrophile dans la membrane permettant le passage des ions chlorures.
Du côté intracellulaire, la protéine CFTR possède un domaine de régulation (domaine R) phosphorylable par la
protéine kinase A. Elle possède également deux domaines de fixation aux nucléotides (ATP) appelés domaines NBD
pour Nucleotide Binsding Domain.
Figure 8:Rôle de la protéine CFTR dans la fluidification du mucus bronchique par
phénomène de transport hydro-électrique.
La protéine CFTR suite à activation par la PKA et fixation d’ATP s’ouvre et permet la
sortie d’ions chlorures selon leur potentiel électrochimique. De plus, CFTR inhibe les
canaux sodiques et le sodium ne rentrant plus s’accumule dans le milieu extracellulaire.
Ces deux phénomènes ont pour conséquence la sortie d’eau par osmose vers le
compartiment à la pression osmotique la plus forte : la lumière de la bronche.
En l’absence de CFTR, les ions chlorures ne sortent plus, les ions sodium rentrent ce
qui fait rentrer l’eau dans le compartiment à plus forte pression osmotique : la cellule. Le
mucus se déshydrate donc et devient plus visqueux.
Figure 6:Méthode de génétique inverse ayant permis de localiser le gène
responsable de la mucoviscidose.
Les méthodes de génétique inverse reposent sur la capacité à évaluer la distance
entre le marqueur de la mucoviscidose (le locus CF) et des marqueurs dont la
position est connue dans le génome. Pour cela, il est nécessaire d’étudier un lot
important de famille attient de mucoviscidose et de calculer des % de
recombinaisons entre le marqueur de position et le marqueur de la maladie. Plus le
pourcentage de recombinaison est élevé plus les deux loci sont éloignés
physiquement dans le génome (probabilité de crossing over forte). Plus le
pourcentage de recombinaison est faible plus les loci sont proches : on dit que les
gènes sont liés.
Il a été remarqué que le locus CF coségrege souvent avec le locus de la paroxonase
(PON) situé sur le chromosome 7. Le gène de la mucoviscidose est donc situé sur le
chromosome 7.
Plus précisément il a été montré qu’il coségrège avec un marquer situé sur le bras
long chromosome 7.
Une fois que la séquence est localisée avec précision, on établit la séquence de la
protéine pour laquelle le gène code. Le gène fait 250 000 paires de bases et contient
27 exons.
Patient « sain »
Patient atteint de mucoviscidose
Figure 11 : Origine de la délation de la phénylalanine 508.
L’alignement des séquences nucléotdiques et protéiques de patients « sains » et atteints de
mucoviscidose met en évidence dans le gène de la mucoviscidose une délétion de 3 paires de
bases : CTT. Cette délétion de trois paires de bases n’entraine pas de décalage du cadre de
lecture (lecture de 3 en 3). En revanche, les trois paires de bases supprimées ne forment pas un
codon : elles sont à cheval sur deux codons successifs. Aussi, le nouveau codon formé par suite à
la délétion est le codon ATT qui, en conséquence de la redondance du code génétique, code
également pour une isoleucine et n’entraine donc qu’une délétion et non pas une délétion et une
mutation.

http://perso.ens-lyon.fr/romain.berardozzi/
Dans tous les cas, CFTR est non-fonctionnel et entraine :
+la séquestration des ions chlorure dans la cellule empêche la sortie d’eau
+l’entrée d’ions sodium dans la cellule suite à la levée d’inhibition par CFTR entraine une
entrée d’eau dans la cellule ce qui déshydrate le mucus.
Les mucus sont donc riches en protéines, glycoprotéines et glucides et pauvre en eau et en
conséquence visqueux expliquant par la suite les symptômes décrit dans le tableau clinique.
La non-fonctionnalité de CFTR réduit également la production d’hypothiocyanate diminuant ainsi les
capacités antimicrobiennes de la muqueuse. De plus, la présence d’un mucus visqueux peu
renouvelé favorise la colonisation de bactéries en biofilms.
Dépistage
Le dépistage de la pathologie peut être fait à plusieurs niveaux (Figure 12) :
+prénatal. Dans le cas des familles à risques (dont un des parents est atteint de la mucoviscidose), un
test génétique permet de vérifier l’existence de porteurs sains et d’établir les risques encourus lors de
la conception d’un enfant. Il y a mise en place d’un conseil génétique en aval de la démarche de
procréation. Il faut noter que les tests disponibles ne permettent pas de s’assurer à 100% de
l’absence de mutations : il s’agit surtout d’une évaluation des risques. En effet, toutes les mutations
ne sont pas facilement testables.
+néonatal. A la naissance, ce dépistage systématique depuis 2002 consiste en un dosage de la
trypsine sanguine par piqure au niveau du talon. La trypsine sanguine est élevée chez les nouveau-
nés dont les canaux pancréatiques sont obstrués. Un test à la sueur (dosage de chlore sudoral) peut
également être réalisé. En effet, les patients atteints de mucoviscidose ont une sueur très salés ([Cl-
]>60mM). Cette méthode de dépistage a été la première au cours de l’histoire à être utilisée.
+dépistage tardif. Suite à l’apparition de symptômes. Les patients dépistés tardivement sont souvent
modérément affectés par la pathologie.
La mucoviscidose, une pathologie nécessitant des traitements thérapeutiques lourds
Réduire les symptômes par des méthodes prophylactiques
En plus des méthodes curatives ci-après, l’apparition des symptômes (notamment les infections
chroniques) peut être limitée par l’application de méthodes prophylactiques (port du masque par le
malade, bonne hygiène corporelle, désinfection de l’environnement).
L’alimentation doit également être surveillé et supplémentée en vitamine afin d’éviter la malnutrition.
Dans les stades avancés, des enzymes pancréatiques peuvent être prescrites.
L’oxygénothérapie permet de compenser le manque d’oxygénation liée à l’obstruction des bronches
L’oxygénothérapie permet d’après des méta-analyses récentes d’améliorer seulement le confort de vie
et non pas la durée de vie de manière statistique. L’oxygénothérapie peut être couplée à une
inhalation de bronchodilatateur permettant d’améliorer les échanges respiratoires au niveau des
surfaces respiratoires.
L’antibiothérapie permet de réduire les infections respiratoires chroniques
L’utilisation d’antibiotiques par voie orale, nébulisation dans les bronches ou par voie veineuse
permet de lutter contre les infections chroniques. L’administration d’antibiotiques à forte dose et de
manière chronique a des effets néfastes chez le patient (problème hépatiques par effet sur les
transaminases notamment). Un risque de résistance aux antibiotiques est également possible
compliquant alors le traitement.
La kinésithérapie permet de réduire l’obstruction des bronches
La kinésithérapie permet d’améliorer l’expectoration bronchique et donc de limiter l’obstruction des
bronches offrant ainsi une meilleure ventilation et une limitation des risques d’infection.
La kinésithérapie permet également de maintenir une mécanique efficace de la ventilation par
renforcement des muscles de la cage thoracique.
 6
7
8
6
7
8
1
/
8
100%