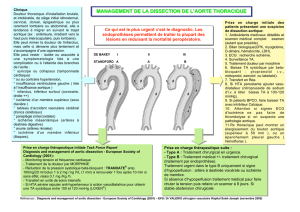
Conséquences métabliques de la reperfusion

ous tenterons de développer dans cette revue les
points essentiels concernant les conséquences méta-
boliques et fonctionnelles de la reperfusion myo-
cardique. Le bien-fondé de la reperfusion est maintenant bien éta-
bli, mais il est des circonstances où cette période post-ischémique
est susceptible d’exercer des effets délétères, qui doivent être envi-
sagés sur les plans spatial et temporel.
NOTION DE RÉVERSIBILITÉ DES ALTÉRATIONS
MÉTABOLIQUES ET DES LÉSIONS ASSOCIÉES À
L’ÉPISODE ISCHÉMIQUE
L’ischémie myocardique peut se définir comme un déséquilibre
entre les besoins tissulaires en oxygène et en substrats nutritifs
et les apports présents dans le sang artériel coronaire. Dans beau-
coup de situations cliniques, l’ischémie myocardique est régio-
nale (localisée à la zone en aval de l’obstruction coronaire). Il
existe en chirurgie cardiaque des situations où l’ischémie myo-
cardique intéresse l’ensemble de l’organe, par exemple, lors de
la mise en route d’une circulation extracorporelle (CEC) ;
l’ischémie est alors globale.
Les nombreuses études expérimentales et cliniques ont précisé
que, si la réversibilité de l’ischémie par la reperfusion intervient
de façon précoce, le tissu préalablement ischémique est capable
de récupérer des caractéristiques fonctionnelles et métaboliques
normales. Plusieurs facteurs tels que la durée de la phase d’isché-
mie, le maintien d’un éventuel débit résiduel et l’importance d’un
possible réseau d’anastomoses et de collatérales vasculaires
conditionnent la cinétique de la récupération post-ischémique.
Une ligature de l’artère circonflexe maintenue pendant quinze
minutes chez le chien provoque une réduction importante du débit
sanguin dans la zone de distribution de l’artère, entraînant des
perturbations significatives des divers paramètres fonctionnels ou
métaboliques. Si le tissu est reperfusé à l’issue de cette période
d’ischémie régionale de quinze minutes, ces paramètres revien-
nent alors rapidement et complètement à leurs valeurs normales.
Un tel processus d’ischémie de courte durée est donc compatible
avec la survie du tissu myocardique, et la reperfusion est alors
susceptible de permettre une totale récupération fonctionnelle.
En revanche, sur le même modèle expérimental, si la ligature
coronaire est maintenue pendant quarante minutes avant que n’in-
tervienne la reperfusion, un phénomène de nécrose se développe
dans la zone sous-endocardique, et la reperfusion n’est plus
capable de stopper cette évolution. Les cellules myocardiques
sont alors entrées dans un processus d’altération irréversible. Les
mécanismes métaboliques impliqués dans le passage de ce point
de non-retour entre une altération réversible et le début du pro-
cessus de nécrose ont fait l’objet de très nombreuses études,
compte tenu de l’importance de cette période de “transition” en
physiopathologie.
MÉTABOLISME ÉNERGÉTIQUE DU MYOCARDE
DANS LES CONDITIONS D’ISCHÉMIE
ET DE REPERFUSION
Le métabolisme du myocarde aboutit à la production de substrats
à liaisons riches en énergie, indispensables aux processus phy-
siologiques à la base du fonctionnement cardiaque : transports
actifs ioniques transmembranaires et contraction cardiaque. Ce
fonctionnement harmonieux nécessite la présence d’oxygène et
de substrats, que la cellule myocardique extrait à partir du sang
coronaire.
Dans les conditions normales, où un flux coronaire adéquat assure
un bon approvisionnement du cœur en substrats et en oxygène,
les différents “combustibles”, qui sont, par ordre préférentiel
d’utilisation, les acides gras (60 %), les lactates (18 %), le glu-
cose (16 %), les acides aminés (3 %) et les corps cétoniques (3 %),
entrent en compétition pour l’oxygène. Il a été clairement démon-
tré que les acides gras estérifiés ou libres ainsi que les triglycé-
rides constituent le substrat préférentiel du muscle cardiaque. En
revanche, le glucose peut, dans certaines circonstances, devenir
le combustible majeur du myocarde, sans toutefois modifier
l’oxydation des acides gras.
MISE AU POINT
La Lettre du Cardiologue - n° 361 - janvier 2003
21
Conséquences métaboliques
de la reperfusion
●L. Rochette, C. Vergely*
*LPPCE, facultés de médecine et de pharmacie, 21000 Dijon.
N

La Lettre du Cardiologue - n° 361 - janvier 2003
22
MISE AU POINT
Le glycogène, quant à lui, n’est pas une source principale d’éner-
gie, excepté dans les situations d’urgence telles qu’une accéléra-
tion subite d’un travail cardiaque ; la libération de ces réserves
glycogéniques dépend surtout du niveau de l’activité du système
sympathique, par l’intermédiaire des catécholamines (noradré-
naline et adrénaline).
Parmi toutes les enzymes qui participent aux réactions de la gly-
colyse, la phosphofructokinase (PFK) est l’enzyme clé, car elle
est très sensible à l’état énergétique des cellules. Elle est régulée
par les taux cellulaires de certains métabolites tels que l’ATP, qui
l’inhibe, et l’ADP, l’AMP et le phosphate inorganique (Pi), qui
l’activent. De même, l’accumulation des protons, donc l’acidose
intracellulaire, réprime l’enzyme, tandis qu’un pH alcalin favo-
rise son activité.
Comme nous l’avons préalablement signalé, les acides gras cir-
culants, qui ont pénétré dans la cellule cardiaque, constituent la
source majeure d’énergie. Sous forme d’acyl CoA, ils passent
dans la matrice mitochondriale par l’intermédiaire du système
carnitine pour y être segmentés, à l’occasion de bêta-oxydations
itératives, en petites molécules d’acétate, qui, comme l’acétate
d’origine glucidique, viennent s’intégrer dans le cycle de Krebs.
Le myocarde est aussi capable de consommer les corps céto-
niques, ainsi que les acides aminés, mais l’importance énergé-
tique de ces apports reste mineure.
S’il existe un élément vasculaire sténosant limitant la circulation
sanguine coronaire, la captation des substrats et l’élimination des
produits du métabolisme cellulaire deviennent problématiques.
L’accumulation de lactate, à laquelle s’ajoutent les métabolites
finaux de la glycolyse, réprime alors la glycolyse au travers de
l’activité d’enzymes telles que la phosphofructokinase, l’inacti-
vation de l’enzyme entraînant secondairement un important ralen-
tissement du flux glycolytique. L’acidification du milieu intra-
cellulaire et la diminution de la synthèse d’ATP vont augmenter
la perméabilité de la membrane cellulaire, ce qui entraîne une
fuite de diverses enzymes cytoplasmiques comme la lactate
déshydrogénase et la créatine kinase, et compromet les transports
actifs. Les conséquences immédiates sont la perturbation des phé-
nomènes électriques et la diminution des niveaux de contraction
myocardique.
Il existe une limite théorique au-delà de laquelle la fonction
cardiaque ne peut pas être restaurée durant la reperfusion
après une ischémie globale. Par des mesures précises, il a été
montré qu’il y avait une chute des taux d’ATP dès les dix pre-
mières secondes d’une ischémie sévère, mais cette chute n’est
pas suffisante pour rendre compte de la réduction importante de
la concentration myocardique. On pourrait alors évoquer la com-
partimentalisation de l’ATP au sein de la cellule.
De l’ATP qui serait localisé dans un “compartiment” membra-
naire spécifique aurait un rôle essentiel dans le maintien de l’in-
tégrité des fonctions membranaires. De nombreuses études réa-
lisées in vivo ont permis de préciser l’incidence du degré et de la
durée de l’ischémie sur l’évolution des teneurs en ATP et en PCr
au niveau du myocarde ischémique. Ainsi, la chute de la PCr est
rapide et importante, alors que le déclin de l’ATP et PCr reflète
la sévérité de l’ischémie myocardique.
ALTÉRATIONS IONIQUES CELLULAIRES
ET DU PH ASSOCIÉES À UN ÉPISODE
ISCHÉMIQUE RÉVERSIBLE
L’ischémie et éventuellement sa réversibilité entraînent des per-
turbations ioniques transmembranaires profondes dont les consé-
quences électrophysiologiques peuvent contribuer à l’apparition
d’arythmies. Ces perturbations sont surtout caractérisées par une
perte de potassium et de magnésium intracellulaires, associée à
une accumulation tissulaire de calcium et de sodium tissulaire.
Le potassium et le sodium
L’ischémie myocardique entraîne une augmentation de la concen-
tration de potassium extracellulaire concomitante d’une aug-
mentation de la glycolyse aérobie, conduisant à une accumula-
tion de lactate, de CO2et de H+. L’existence d’une relation entre
arythmies et augmentation de la concentration de potassium extra-
cellulaire a été rapportée. Cette concentration élevée de potas-
sium extracellulaire provoque une dépolarisation des cellules
ischémiques. Elle peut également induire une inhibition du cou-
rant sodique rapide et déterminer une activité ectopique. L’aug-
mentation de la concentration extracellulaire de potassium durant
l’ischémie ou la reperfusion peut s’expliquer par une inhibition
de la pompe Na+-K+. La fuite massive de potassium du com-
partiment intracellulaire vers le compartiment extracellulaire ne
peut provenir que d’une augmentation de la perméabilité de la
membrane cellulaire qui fait suite à l’ischémie.
In vitro, de nombreuses études ont précisé que l’intensité des
arythmies d’ischémie et de reperfusion est influencée par la
concentration en potassium du milieu de perfusion ; un milieu
hypokaliémique favorise la survenue des fibrillations ventricu-
laires durant les phases d’ischémie et de reperfusion.
Comme nous l’avons précisé précédemment, l’ischémie est sui-
vie d’une réduction des concentrations tissulaires en ATP et phos-
phocréatine, associée à une acidification intracellulaire liée à l’ac-
célération de la glycolyse anaérobie. Il est admis que la diminution
du pH intracellulaire provoque l’activation de l’échangeur Na+/H+,
accompagnée d’une augmentation transitoire des concentrations
intracellulaires de sodium.
La reperfusion va être induite, alors que le myocarde “ischémique”
est un tissu où les teneurs en substrats énergétiques sont très abais-
sées et où le pH intracellulaire s’est acidifié. Il sera très important
de considérer les conditions de reperfusion pour comprendre
l’adaptation du myocarde à cette nouvelle phase. Une charge de
travail imposée au myocarde qui serait supérieure à ses possibili-
tés énergétiques viendrait exacerber son dysfonctionnement.
Le calcium
Il existe un gradient de concentrations entre le cytosol et les orga-
nites intracellulaires susceptibles de “stocker” du calcium. Le cal-
cium est inégalement distribué à l’intérieur des cellules. Les mou-
vements du calcium à travers le sarcolemme impliquent des
transferts d’ions à travers des canaux sensibles aux différences
de potentiel transmembranaire et la mise en jeu de transporteurs.

La Lettre du Cardiologue - n° 361 - janvier 2003
23
Si des incertitudes demeurent encore sur l’intensité de l’aug-
mentation des taux de calcium au cours de la séquence ischémie-
reperfusion, des travaux ont permis de mesurer une augmenta-
tion du calcium libre cytosolique au cours des dix premières
minutes d’une ischémie totale sur le cœur, ainsi que des modifi-
cations de la distribution intracellulaire de cet ion au cours de la
phase de reperfusion. L’élévation du calcium est suivie d’une acti-
vation de nombreuses enzymes (protéases, lipases), d’une dimi-
nution de l’ATP liée à l’activation de nombreuses ATPases et
d’une inhibition de phosphorylations oxydatives mitochondriales.
Les mitochondries sont alors capables d’accumuler de grandes
quantités de calcium. L’accumulation intramitochondriale de cal-
cium sous forme d’inclusions denses aux électrons a été mise en
évidence. Par ailleurs, une augmentation des concentrations cyto-
soliques des ions H+pourrait déplacer des ions Ca2+ de ses sites
de liaisons. De telles interactions seraient importantes à consi-
dérer dans des situations telles que l’ischémie, qui est associée à
une diminution du pH intracellulaire.
Le magnésium
Le magnésium, second cation intracellulaire, joue un rôle dans
un grand nombre de réactions biochimiques. Au niveau cardiaque,
le magnésium influe sur l’activité sinusale et sur la conduction
auriculoventriculaire de l’excitation. Il diminue la contractilité
du muscle cardiaque, mais il est indispensable au déclenchement
des contractions normales. Sur le cœur ischémique, la perte de
magnésium qui s’établit durant l’ischémie est associée au déve-
loppement d’atteintes tissulaires qui perturbent le processus de
récupération ultérieure. Un effet protecteur du magnésium a été
décrit avec une solution cardioplégique en contenant des
concentrations élevées.
RÔLE DU SYSTÈME NERVEUX SYMPATHIQUE
DANS LES MODIFICATIONS FONCTION-
NELLES CARDIAQUES ASSOCIÉES À LA
SÉQUENCE ISCHÉMIE-REPERFUSION
L’ischémie entraîne une augmentation des teneurs tissulaires
en AMP cyclique dans le myocarde ischémique. Des niveaux
élevés d’AMP cyclique sont toutefois retrouvés dans certaines
études, aussi bien dans les zones ischémiées que dans les zones
non ischémiées. L’élévation des taux tissulaires dans le myo-
carde ischémique peut être interprétée comme étant secondaire
à une hyperactivité sympathique et à la stimulation des récep-
teurs bêta-adrénergiques. Il est d’ailleurs important de signa-
ler qu’au niveau du myocarde qui ne subit pas une ischémie,
une adaptation métabolique et fonctionnelle se met en place
très rapidement. La genèse des troubles graves du rythme car-
diaque qui s’installent durant les phases d’ischémie et/ou de
reperfusion est, selon de nombreux auteurs, associée à l’aug-
mentation des teneurs tissulaires d’AMP cyclique. Il a été mon-
tré, d’autre part, qu’une perfusion de dibutyryl AMP cyclique
abaisse le seuil de fibrillation, effet qui est également retrouvé
après blocage de la dégradation de l’AMP cyclique par inhi-
bition de la phosphodiestérase. Au niveau du cœur ischémique,
les modifications de la cinétique de libération et du métabolisme
des catécholamines jouent un rôle important dans la genèse des
arythmies induites par l’ischémie ou la reperfusion (1, 2). L’in-
tervention des catécholamines dans le déclenchement de ces
arythmies a été le plus souvent évoquée dans le contexte d’une
situation de stress mettant en jeu l’ensemble du système sympa-
thique, sans que soit précisé le rôle spécifique des catécholamines
myocardiques.
RÔLE DES RADICAUX LIBRES ET DE LA PER-
OXYDATION LIPIDIQUE DANS LES ALTÉRATIONS
CELLULAIRES INDUITES PAR L’ISCHÉMIE ET/OU
LA REPERFUSION
(figure 1)
Les processus physiopathologiques impliqués dans les altérations
cellulaires induites par l’ischémie myocardique réversible restent
complexes. Au cours de ces dernières années, les modifications
moléculaires ont été étudiées pour tenter d’expliquer les condi-
tions d’installation et les niveaux des altérations fonctionnelles
et morphologiques. Dans ce cadre, le rôle des radicaux libres a
été évoqué (3). Les radicaux libres ont été impliqués en biologie
après les travaux de McCord et Fridovich, qui mirent en évidence
la formation de l’anion superoxyde au cours d’une oxydation
enzymatique et l’enzyme superoxyde dismutase. Les études ulté-
rieures ont montré que l’anion superoxyde mais également
d’autres espèces radicalaires sont générés au cours de nombreuses
MISE AU POINT
Ca++ intracellulaire
K+ intracellulaire
Stimulation
adrénergique
Libération de la noradrénaline
ATP
AMP Perturbation
des fonctions
mitochondriales
Délocalisation
du fer
Adénosine
Inosine
I
N
C
I
D
E
N
C
E
S
F
O
N
C
T
I
O
N
N
E
L
L
E
S
Actions
membranaires Altérations
membranaires
Hydroperoxydes
Phospholipase A2
Phospholipides
Endoperoxydes MDA
Acide arachidonique
Hypoxanthine
Xanthine oxydase
Radicaux libres
Prostacycline Thromboxane Leucotriène
Cyclo-oxygénase
Lipo-oxygénase
Ischémie-Reperfusion
Figure 1. Schéma des différents événements métaboliques associés à la séquence ischémie-
reperfusion.

La Lettre du Cardiologue - n° 361 - janvier 2003
24
réactions biochimiques intracellulaires et “éliminés” par diffé-
rents systèmes de protection. Dans certaines conditions patholo-
giques, ces systèmes de défense peuvent être saturés ; les radi-
caux libres sont alors capables d’altérer les constituants cellulaires
et d’induire des lésions graves. De nombreux travaux suggèrent
qu’au cours des processus d’ischémie et de reperfusion, la pro-
duction de radicaux libres et la défaillance progressive des sys-
tèmes cellulaires de défense chargés de leur élimination pour-
raient contribuer au développement des altérations cellulaires qui
caractérisent cette séquence (4).
La toxicité des radicaux libres semble en fait être en grande par-
tie liée au radical hydroxyle, très instable et très réactif. Son pou-
voir oxydant très puissant lui permet de réagir sur son site de pro-
duction avec pratiquement tous les substrats organiques :
amino-acides, sucres, lipides, acides organiques, acides
nucléiques, etc. Son espace de distribution est faible du fait de sa
grande réactivité. En revanche, il est à l’origine d’une cascade de
réactions radicalaires (5, 6).
Le rôle des métaux de transition dans la cascade des réactions
radicalaires est très complexe, car un grand nombre de réactions
sont possibles entre les métaux oxydés ou réduits et les diffé-
rentes formes activées de l’oxygène. Le fer et le cuivre cataly-
sent la dégradation du peroxyde d’hydrogène, en présence ou non
d’anions superoxyde, en radical hydroxyle au cours de la réac-
tion de Fenton et du cycle de Haber-Weiss.
La capacité des métaux chélatés à former des complexes stables
avec l’oxygène, le superoxyde ou d’autres espèces réduites de
l’oxygène est souvent ignorée dans les milieux biologiques, mais
semble importante. Ce phénomène pourrait permettre une diffu-
sion des radicaux libres et l’induction de dommages cellulaires
à une certaine distance de leur site de production. Cela pourrait
expliquer le fait que certains phénomènes oxydatifs ne sont pas
prévenus par les piégeurs classiques de radicaux libres ; la com-
plexation “métal-forme réduite de l’oxygène” pourrait en effet
protéger les radicaux oxygénés des piégeurs endogènes.
Divers mécanismes concourent à une augmentation de la pro-
duction de radicaux libres durant les phénomènes d’ischémie et
de reperfusion cardiaque (7). Durant l’ischémie, l’apport en oxy-
gène réduit perturbe le fonctionnement de la chaîne respiratoire.
La réduction monovalente de l’oxygène est favorisée, ainsi que
la libération de composés tels que les flavoprotéines, les ubiqui-
nones, ce qui induit une production de radicaux libres. Il est alors
facile d’envisager qu’une altération du métabolisme oxydatif de
la cellule s’installe, ce qui favorise la détérioration des mito-
chondries, créant ainsi un cercle vicieux qui précipite le dys-
fonctionnement cellulaire.
Parmi les autres processus mis en jeu et qui participent à la géné-
ration des espèces radicalaires, l’activation des oxydases occupe
une place majeure ; ainsi, la xanthine oxydase constitue une
source de radicaux libres. Dans les tissus, l’enzyme est présente
en grande partie sous la forme xanthine déshydrogénase, qui ne
peut réduire l’oxygène moléculaire, mais une activité xanthine
oxydase existe cependant. Durant l’ischémie, la conversion de la
xanthine déshydrogénase en xanthine oxydase peut être réalisée
par une protéase activée par l’accumulation de calcium cytoso-
lique ou par l’oxydation des groupements thiols. Les NAD(P)H
oxydases se trouvent également activées, soit de manière directe,
soit de manière indirecte (par exemple, après stimulation de récep-
teurs tels que les récepteurs à l’angiotensine II [8]).
Rappelons également que l’acidose qui se développe au cours de
l’ischémie peut favoriser :
✔la formation de la forme protonée du superoxyde, avec accé-
lération de la dismutation du superoxyde en peroxyde d’hydro-
gène ;
✔la libération du fer de complexes inactivateurs et la formation
de radical hydroxyle par diverses réactions, dont les réactions de
Haber-Weiss et de Fenton et la production de radicaux libres.
L’activation des phospholipases par l’accumulation de calcium
cytosolique peut induire une dégradation des phospholipides
membranaires et la libération d’acide arachidonique, ce qui sti-
mule le métabolisme de l’acide arachidonique et la formation
concomitante de radicaux libres par la cyclo-oxygénase.
Une production radicalaire importante au cours d’un épisode
ischémique suivi d’une reperfusion est liée au processus inflam-
matoire (9). Le processus inflammatoire a fait l’objet de très nom-
breux travaux cherchant à définir des stratégies qui inhiberaient
la voie du complément (10).
In vivo, les leucocytes qui envahissent la zone ischémiée consti-
tuent une source extracellulaire de radicaux libres selon les méca-
nismes décrits précédemment. L’ischémie et l’inflammation agis-
sent en synergie. La production de radicaux libres peut induire la
formation de facteurs chémotactiques – dont certains sont déri-
vés de la peroxydation lipidique – qui provoquent un afflux de
neutrophiles activés par les composés issus de la dégradation cel-
lulaire induite par les radicaux libres. Par formation d’agrégats,
ils vont modifier la microcirculation, ce qui exacerbe le phéno-
mène ischémique. Par ailleurs, l’augmentation de la perméabi-
lité vasculaire induit la formation d’un œdème qui perturbe éga-
lement la microcirculation et favorise le processus ischémique.
La lyse des hématies présentes dans les capillaires libère des déri-
vés du fer, qui favorisent, par auto-oxydation ou catalyse de la
réaction de Fenton, la production de radicaux libres, et partici-
pent ainsi au développement d’altérations cellulaires (11).
Les radicaux libres oxygénés sont des agents chémotactiques pour
les plaquettes et les PNN, qui adhèrent à l’endothélium et for-
ment des thrombi, et constituent une composante supplémentaire
des effets délétères des radicaux libres oxygénés à la reperfusion.
L’ensemble de ces altérations pourrait être à l’origine du “stun-
ning” myocardique et, indirectement, du phénomène de “no-
reflow”. Le NO est également un radical libre, mais il tient une
place à part dans la séquence ischémie-reperfusion. Il existe une
production basale de NO par les cellules endothéliales jouant un
rôle physiologique de vasodilatateur. Après une séquence isché-
mie-reperfusion, la relaxation endothéliale-dépendante des artères
coronaires à la plupart des agonistes vasoactifs est altérée, bien
que les propriétés contractiles des cellules musculaires lisses
soient intactes. La dysfonction endothéliale due à la reperfusion
pourrait alors en partie expliquer le stunning microvasculaire et
contribuer au vasospasme coronaire. Ce phénomène pourrait être
MISE AU POINT

La Lettre du Cardiologue - n° 361 - janvier 2003
25
attribué à une diminution du NO produit et/ou à une diminution
de sa disponibilité par le biais de sa réaction avec l’anion super-
oxyde. La réduction de la disponibilité du NO serait alors délé-
tère pour le myocarde. En effet, le NO est vasodilatateur, et il pos-
sède la propriété d’inhiber l’adhésion endothéliale des
polynucléaires neutrophiles (PNN) et des plaquettes ; la perte de
cette propriété favoriserait une vasoconstriction, ainsi que des
phénomènes d’agrégation et d’adhésion des PNN et des pla-
quettes. La contraction des vaisseaux intéressés pourrait contri-
buer à la survenue du spasme vasculaire ou à la diminution du
débit coronaire, perpétuant ainsi l’ischémie. Par ailleurs, il semble
actuellement admis que le peroxynitrite formé possède lui aussi
des propriétés cytotoxiques ; il pourrait alors jouer un rôle majeur
dans l’initiation des lésions post-ischémiques.
CONCLUSION
Dans le cadre de la réflexion globale sur les liens qui unissent le
processus inflammatoire, la migration des cellules de défense et
le devenir des cellules constituant le myocarde, un volet nouveau
de la protection myocardique passe par la cible des métallopro-
téinases de la matrice extracellulaire (MMP) (12). Les neutro-
philes qui infiltrent le tissu constituent une source de MMP. Il est
intéressant également de rappeler que des travaux récents (13)
ont démontré que MMP-2 jouait un rôle dans l’adaptation de l’en-
dothélium vasculaire à la séquence hypoxie-réoxygénation, et que
le peroxynitrite produit au cours de la reperfusion pouvait acti-
ver les MMP (14). L’avenir dira si l’inhibition de l’activité des
MMP constitue réellement une voie de protection efficace du
myocarde comparativement à celles qui sont actuellement démon-
trées (15). ■
Bibliographie
1. Schomig A. Catecholamines in myocardial ischemia, systemic and cardiac
release. Circulation 1990 ; 82 : 13-22.
2. Seyfarth M, Richardt G, Mizsnyak A, Kurz T, Schomig A. Transcient ischemia
reduces norepinephrine release during sustained ischemia. Neural preconditio-
ning in isolated rat heart. Cir Res 1996 ; 78 (4) : 573-80.
3. Wems SW, Lucchesi BR. Free radicals and ischemic tissue injury. Trends
Pharmacol Sci 1990 ; 11 (4) : 161-6.
4. Vergely C, Maupoil V, Benderitter M, Rochette L. Influence of the severity of
myocardial ischemia on the intensity of ascorbyl-free radical release and on post-
ischemic recovery during reperfusion. Free Radic Bio Med 1998 ; 24 (3) : 470-9.
5. Buettner GR, Jurkiewicz BA. Catalytic metals, ascorbate and free radicals :
combinations to avoid. Radiat Res 1996 ; 145 : 532-41.
6. Halliwell B. The chemistry of free radicals. Toxicol Ind Health 1993 ; 9 (1-2) :
1-21.
7. Hearse D, Bolli R. Reperfusion-induced injury : manifestations, mechanisms
and clinical relevance. Cardiovasc Res 1992 ; 26 : 101-8.
8. Griendling KK, Sorescu D, Ushio-Fukai M. NAD(P)H oxidase : role in car-
diovascular biology and disease. Circ Res 2000 ; 86 (5) : 494-501.
9. Babior BM. Phagocytes and oxidative stress. Am J Med 2000 ; 109 (1) : 33-44.
10. Monsinjon T, Richard V, Fontaine M. Complement and its implications in car-
diac ischemia/reperfusion : strategies to inhibit complement. Fundam Clin
Pharmacol 2001 ; 15 (5) : 293-306.
11. Clermont G, Vergely C, Lahet JJ et al. Systemic free radical activation is a
major event involved in myocardial oxidative stress related to cardiopulmonary
bypass. Anesthesiology 2002 ; 96 : 80-7.
12. D’Armiento J. Matrix metalloproteinase disruption of the extracellular
matrix and cardiac dysfunction. Trends Cardiovasc Med 2002 ; 12 : 97-101.
13. Ben-Yosef Y, Lahat N, Shapiro S, Bitterman H, Miller A. Regulation of endo-
thelial matrix metalloproteinase-2 by hypoxia/reoxygenation. Circ Res 2002 ; 90
(7) : 784-91.
14. Wang W, Sawicki, Schulz R. Peroxynitrite-induced myocardial injury is
mediated through matrix metalloproteinase-2. Cardiovasc Res 2002 ; 53 (1) :
165-74.
15. Wang QD, Pernow J, Sjöquist PO, Ryden L. Pharmacological possibilities
for protection against myocardial reperfusion injury. Cardiovasc Res 2002 ; 55 :
25-37.
MISE AU POINT
1
/
5
100%