VE-BASKET resume

Chers Amis,
Chers collègues,
Comme vous le savez, la présence d’une mutation activatrice BRAF V600E est présente dans
certains mélanomes. Les traitements qui ciblent cette protéine activée ont démontré leur efficacité
dans les mélanomes métastatiques et le Vémurafénib a obtenu une AMM dans cette indication.
Nous ne savons pas si cette mutation a le même potentiel « thérapeutique » dans d’autres cancers,
dans lesquels elle peut être présente soit fréquemment, soit de façon plus rare (cf infra)
Plusieurs études tentent de répondre à cette question :
-
Pour les cancers bronchiques, la recherche de BRAF est recherchée systématiquement (1 à
2% d’incidence attendue) et les dossiers de vos patients qui présenteraient cette anomalie
peuvent être discutés à la RCP « moléculaire » qui se tient tous les lundis à 14 heures dans le
service de Pneumologie et que nous animons avec Julien Mazieres. Les patients « BRAF »
peuvent entrer dans une étude dédiée organisée par Julien et son équipe.
-
Pour ce qui concerne les cancers du colon (5 à 15% d’incidence attendue), des études sont
accessibles en phase I ou IB à l’ICR (les places sont un peu plus contingentées). Vous
pouvez me contacter ou contacter l’équipe pour avoir plus de renseignements.
-
Pour toutes les autres formes de cancers dont voici la liste, un programme de
recherche est disponible dans :
o
Cancers de l’ovaire (30% de mutation BRAF attendue)
o
Cholangiocarcinome (15 à 20%)
o
Cancer du sein (2%)
o
Cancer de prostate (4 %)
o
Myelome (2%)
o
« tumeurs rares » : cancers des glandes salivaires par exemple…
Il existe actuellement dans 8 centres en France (dont l’ICR) la possibilité d’avoir accès au
Vémurafenib dans une phase II « exploratoire » dans laquelle toutes ces cohortes sont représentées.
La plateforme régionale de biologie moléculaire se tient à votre service pour réaliser la recherche de
mutations. Si vous renseignez la demande via une fiche « biologie moléculaire » habituelle en
signalant simplement l’item « phase I ». Nous nous chargeons de financer les frais engagés par votre
demande via le(s) programme(s) de recherche.
Janick Selves travaille actuellement avec Oncomip dans l’objectif de vous fournir rapidement une fiche
« dédiée ». En attendant, utiliser votre fiche habituelle de demande est possible.
L’idée est de permettre à vos patients d’avoir accès à cette évaluation moléculaire. Bien entendu,
nous parlons d’une étude de phase II dont l’objectif est de cherche une première « preuve de
concept ». Les patients doivent répondre à des critères de bon état général, comme dans la plupart
des études cliniques. Les critères sont ici néanmoins un peu moins « durs » que dans les phases I.
L’étude restera ouverte pendant les 6 prochains mois, ensuite…cela dépendra de la vitesse
d’inclusion dans les autres centres en France et dans le monde…
N’hésitez pas à nous contacter si vous souhaitez le synopsis ou des renseignements plus précis.
J’espère que cette ouverture d’étude permettra de rendre service à certains de vos patients et qu’elle
contribuera à continuer de structurer la circulation des informations biologiques en Midi Pyrénées.
Nous travaillons à cela afin conserver au sein d’Oncomip des capacités de répondre au défis de la
prise en charge des cancers à l’avenir.
Amicalement
Cordialement
Confraternellement
Jean-Pierre Delord

HO ; VE-BASKET ; N°1 version BEC CONFIDENTIEL – NE PAS DIFFUSER page 1 sur 8
VE-BASKET
Étude de phase II, en ouvert, évaluant l’efficacité du vemurafenib
chez des patients atteints d’un cancer porteur de la mutation BRAF
V600.
MO28072
12GENE08 ; IP : Pr J.P. DELORD ; Promoteur : ROCHE ; ouvert à l’ICR le 14/11/2012
ARC en charge de l’étude à l’ICR : Harmonie OULIE, Poste 2732.
Résumé rédigé à partir de la version A du protocole du 22 août 2012.
Version BEC n° 1.
Etude de Phase II, multicentrique, en ouvert, à 8 cohortes.
1.
Objectif Principal
Evaluer l’efficacité du vemurafenib par le taux de réponse à 8 semaines de
traitement, évalué par l’investigateur selon les critères RECIST 1.1 ou IMWG
(International Myeloma Working Group) chez des patients atteints d’un cancer
porteur d’une mutation BRAF V600.
2. Objectifs Secondaires
Evaluer la sécurité et la tolérance du vemurafenib dans cette population
de patients.
Evaluer dans les tumeurs solides et le myélome multiple : le taux de
réponse globale, le bénéfice clinique [réponse complète ou réponse
complète stringente, réponse partielle ou très bonne réponse partielle et
maladie stable] du vemurafenib, la durée de la réponse, le délai de
réponse, le temps jusqu’à progression, la survie sans progression et la
survie globale.
Evaluer le test de diagnostic compagnon (CoDx) Roche cobas® 4800
BRAF V600 pour la détection de mutations BRAF V600 dans des
échantillons tumoraux.
3. Critères d’Inclusion Exhaustifs
Pour les tumeurs solides uniquement : (hors mélanome et cancer
papillaire de la thyroïde)
Cancer confirmé histologiquement (hors mélanome et cancer papillaire
de la thyroïde), porteur d’une mutation BRAF V600 et réfractaire au
traitement standard ou pour lequel le traitement standard ou curatif
n’existe pas ou n’est pas approprié selon l’investigateur.
Tumeur mesurable selon les critères RECIST 1.1.
Fonction hématologique satisfaisante, analyses réalisées dans les 7
jours avant la première prise de vemurafenib :
Nombre absolu de neutrophiles (ANC) ≥ 1,5 x 10
9
/L.
Numération plaquettaire ≥ 100 x 10
9
/L.
Pour les myélomes multiples (MM) uniquement : (MM caractérisé par un
plasmocyte solitaire extra-médullaire ou osseux)
Patients avec un diagnostic de MM confirmé, porteur d’une mutation
BRAF V600.
Patients ayant reçu au moins une première ligne de traitement pour un
MM. Une ligne de traitement est un traitement séquentiel, sans
interruption en cas de réponse et progression ultérieure.
Si le patient est traité par radiothérapie locale (avec / sans
administration concomitante à des corticoïdes pour contrôler la douleur
ou gérer des compressions des racines nerveuses / de la moelle
épinière) : 2 semaines doivent s’être écoulées depuis la dernière séance
de radiothérapie, qui est recommandée sur un champ limité. Les
patients ayant besoin d’une radiothérapie concomitante doivent retarder
leur entrée dans l’étude jusqu’à ce que la radiothérapie soit terminée et
que 2 semaines se soient écoulées depuis la dernière séance.
Patients atteints d’un MM en rechute et / ou réfractaire avec une
maladie mesurable, définie comme une maladie qui peut être mesurée
soit par une analyse sérique ou urinaire de l’immunoglobuline
monoclonale, soit par la recherche de chaînes légères libres (FLC)
sériques pour un des paramètres suivants :
Protéine M sérique > 0,5 g/dL
Protéine M urinaire > 200 mg / 24h
Taux de FLC impliquées > 10 mg/dL (> 100 mg/L) si le ratio de
FLC est anormal.
Fonction hématologique satisfaisante, analyses réalisées dans les 7
jours avant la première prise de vemurafenib :
Nombre absolu de neutrophiles (ANC) ≥ 1,0 x 10
9
/L.
Numération plaquettaire ≥ 50 x 10
9
/L.

HO ; VE-BASKET ; N°1 version BEC CONFIDENTIEL – NE PAS DIFFUSER page 2 sur 8
Pour tous les patients (tumeurs solides et MM) : consentement éclairé écrit
obtenu avant le début de toute procédure spécifique à l’étude.
En cas de grossesse chez la partenaire d’un patient : signature d’un
consentement AVANT notification de la grossesse.
Homme ou femme âgé(e) de 18 ans ou plus.
Indice de performance ECOG de 0 à 2.
Tous les effets indésirables dus au traitement antérieur systémique ou
local le plus récent doivent être résolus (sauf pour l’alopécie).
Patients aptes à avaler des comprimés.
Fonction hématologique satisfaisante, analyses réalisées dans les 7
jours avant la première prise de vemurafenib :
Hémoglobine ≥ 9 g/dL
Créatinine sérique ≤ 1,5 fois la limite normale supérieure (LNS)
ou clairance de la créatine (ClCr) > 50 mL/min selon la formule
de Cockroft-Gault
ASAT et ALAT ≤ 2,5 x LNS (≤ 5 x LNS si résultats liés à une
atteinte hépatique primaire ou métastatique)
Bilirubine sérique ≤ 1,5 x LNS
Phosphatase alcaline ≤ 2,5 x LNS (≤ 5 x LNS si résultat lié à la
tumeur).
Test de grossesse sérique négatif dans les 7 jours précédant la
première administration chez les femmes en pré ménopause. Les
femmes qui ne sont pas en âge de procréer peuvent être incluses sans
réaliser de test de grossesse si elles ont eu une stérilisation chirurgicale
ou si elles sont en post-ménopause depuis ≥ 1 an.
Les hommes fertiles et les femmes en âge de procréer doivent utiliser
une méthode de contraception efficace pendant le traitement et au
moins 6 mois après la fin du traitement. Les méthodes efficaces de
contraception sont définies comme ayant un faible taux d’échec
(inférieur à 1% par an) quand elles sont utilisées systématiquement et
correctement (ex : implants, contraceptifs injectables, contraception
oestro-progestative orale, dispositifs intra-utérins). A l’appréciation de
l’investigateur, l’abstinence sexuelle totale peut être une méthode
acceptable pour des cas où le style de vie du patient ou de la patiente
garantit la compliance. Une abstinence périodique (ex : méthodes du
calendrier, de l’ovulation ou post-ovulation, sympto-thermique) et le
retrait ne sont pas des méthodes de contraception acceptables.
Absence de tout facteur psychologique, familial, sociologique ou
géographique susceptibles de faire obstacle à l’observance du protocole
et du calendrier de suivi. Ces points doivent être discutés avec le patient
avant sa participation à l’étude.
Patient affilié à un régime / bénéficiaire de la sécurité sociale.
4. Critères de non-Inclusion Exhaustifs
Mélanome, cancer papillaire de la thyroïde et hémopathies (sauf MM).
Maladie maligne non contrôlée (une maladie chronique ou à un stade
précoce sera acceptée si elle est contrôlée et ne nécessite pas de
traitement ou d’intervention).
MM pour lesquels la seule mise en évidence de cellules plasmocytaires
est un plasmocytome solitaire extra-médullaire ou osseux.
Présence de métastases du système nerveux central, actives ou non
traitées. Les patients avec des métastases cérébrales sont éligibles s’ils
sont asymptomatiques, non traités par corticothérapie et sans
progression cérébrale de la maladie depuis ≥ 2 mois.
Antécédent de méningite carcinomateuse.
Tout traitement anticancéreux concomitant (ex : chimiothérapie, autre
thérapie ciblée, autre traitement expérimental,...) autre que celui
administré dans cette étude.
Hypersensibilité connue au vemurafenib ou tout autre BRAF inhibiteur.
Traitement antérieur par un inhibiteur de BRAF ou de MEK (un
traitement antérieur par sorafenib est autorisé).
Femmes enceintes ou allaitantes.
Nausées et vomissements réfractaires, malabsorption, dérivation biliaire
externe ou résection intestinale importante qui empêcherait une
absorption suffisante.
Survenue de l’un des événements suivants dans les 6 mois précédant la
première administration du vemurafenib : infarctus du myocarde, angine
de poitrine sévère / instable, insuffisance cardiaque congestive
symptomatique, AVC ou accident ischémique transitoire.
Embolie pulmonaire dans les 30 jours précédant la première
administration du vemurafenib.
Hypertension non contrôlée par les traitements standards dans les 30
jours précédant la première administration du vemurafenib.
Antécédent ou présence de troubles du rythme ventriculaire ou
auriculaire cliniquement significatifs ≥ grade 2 (NCI CTCAE v4.0).
Intervalle QT corrigé (QTc) ≥ 450 msec à la visite de baseline ou
antécédent de syndrome du QT long congénital, ou électrolytes
anormaux non corrigés.
Affection médicale non contrôlée (telle qu’une infection nécessitant un
traitement antibiotique intraveineux).
Autre affection médicale ou psychiatrique sévère, aiguë ou chronique,
ou anomalies biologiques pouvant augmenter le risque lié à la
participation à l’étude ou à la prise du vemurafenib ou pouvant interférer
avec l’interprétation des résultats de l’étude, selon l’appréciation de
l’investigateur.

HO ; VE-BASKET ; N°1 version BEC CONFIDENTIEL – NE PAS DIFFUSER page 3 sur 8
Patients ne souhaitant pas utiliser une contraception efficace.
Incapacité à respecter les autres exigences du protocole.
5. Modalités du Traitement
L’étude comportera 8 cohortes de 7 patients chacune pour les cancers suivants :
Cohorte 1. Cancer bronchique non à petites cellules (CBNPC)
Cohorte 2. Cancer de l’ovaire
Cohorte 3. Cancer colorectal
Cohorte 4. Cholangiocarcinome / Cancer des voies biliaires
Cohorte 5. Cancer du sein
Cohorte 6. Cancer de la prostate
Cohorte 7. Myélome multiple (MM)
Cohorte 8. Tumeurs solides autres que celles ci-dessus.
Les patients inclus recevront oralement et en continu 960 mg de vemurafenib,
deux fois par jour.
Dans le cas où le statut BRAF V600 du patient n’est pas connu, il est
possible de le déterminer :
Soit par biopsie fraîche après signature du consentement
Soit sur un bloc / lame / ADN tumoral archivé : l’analyse est
réalisable avant la signature du consentement (information seule
du patient de l’utilisation de sa tumeur archivée).
Un consentement optionnel pour un test BRAF rétrospectif (relecture
centralisée) et archivage de l’ADN durant 5 ans est à proposer au patient.
Phase de screening : 28 jours, à partir de la date de signature du CE.
Phase de traitement : Un cycle de traitement = 28 jours.
Phase de suivi : cf. 11.
6. Recommandations dans l’administration du produit
Il sera remis aux patients 2 flacons de 120 comprimés de 240 mg chacun (stock
= 30 jours), à conserver à 25°C ou moins (au réfrigérateur l’été si besoin).
La prise : 4 comprimés le matin et 4 comprimés 12 heures plus tard, soit
dans la soirée, pour un total de 1920 mg par jour (= 960 mg BID).
Les 2 flacons dispensés devront être retournés à la visite suivante (vides ou
avec de comprimés).
Dans tous les cas, le premier flacon entamé doit toujours être terminé avant
d’utiliser le second.
Cas particulier : lors de la première dispensation, 3 flacons (et non 2) seront
fournis aux patients. Un de ces 3 flacons sera à conserver jusqu’à péremption
chez le patient (flacon de secours en cas de visite reportée, perte d’un flacon,…)
Le vemurafenib est à avaler avec un verre d’eau (ne pas croquer ni
écraser) tous les jours, de la même manière et à la même heure.
La prise peut s’effectuer pendant ou hors des repas, mais il faut
conserver ce choix pendant toute l’étude.
En cas de vomissement d’un comprimé, ne pas prendre un autre
comprimé.
Si le comprimé n’est pas pris à l’heure théorique :
Prendre la dose oubliée s’il reste 4 heures ou plus avant l’heure
de la prochaine prise. La dose suivante sera prise à la même
heure qu’habituellement.
Ne pas prendre la dose oubliée s’il reste moins de 4 heures avant
la dose suivante. Noter cette prise ratée dans le carnet patient.
7. Durée du Traitement
Les patients inclus prendront le traitement jusqu’à progression de la maladie
(évaluée par l’investigateur), toxicité inacceptable, retrait du consentement,
violation de protocole mettant en danger la sécurité du patient, décès, raisons
jugées valables par l’investigateur, ou arrêt de l’étude par le promoteur.
Si une progression tumorale est suspectée mais, si de l’avis de l’investigateur, la
poursuite du vemurafenib reste bénéfique pour le patient, le traitement à l’étude
pourra être continué après discussion avec le promoteur.
8. Evaluation tumorale
Tumeurs solides :
La réponse sera évaluée grâce aux critères RECIST 1.1.
Les évaluations tumorales auront lieu au screening, toutes les 8
semaines (2 cycles) après le début du vemurafenib durant la période de
traitement et lors de la visite de fin de traitement. Une fenêtre de 5 jours
est autorisée.
L’évaluation tumorale sera réalisée via IRM ou TAP-scan : la même
technique et le même relecteur sont requis tout au long de l’étude.
Nb : pour les patients répondeurs (réponse partielle ou réponse complète), les
changements dans les mesures de la tumeur doivent être confirmés par
une nouvelle évaluation au minium 4 semaines après l’évaluation où la
première réponse a été vue.

HO ; VE-BASKET ; N°1 version BEC CONFIDENTIEL – NE PAS DIFFUSER page 4 sur 8
Myélomes multiples :
La réponse sera évaluée grâce aux critères IMWG (Annexe 1).
Les évaluations tumorales auront lieu au screening, 8 semaines après le
début du vemurafenib (J1C3), puis tous les 4 semaines (à chaque J1) et
lors de la visite de fin de traitement.
L’évaluation et la confirmation d’une réponse complète sera faite une
seule fois sur une ponction (myélogramme) / biopsie médullaire, après 2
évaluation consécutives négatives.
Nb : les patients seront considérés comme répondeurs lorsqu’une réponse
complète, une réponse complète stringente, une très bonne réponse partielle ou
une réponse partielle est décrite lors de 2 évaluations consécutives. Le
myélogramme peut être programmé à ce moment.
9. Autres évaluations
Carcinomes épidermoïdes cutanés :
Dans ce cas, il faut prévoir une consultation au screening, une consultation
mensuelle durant la phase de traitement et 6 mois après l’arrêt du
traitement avec le dermatologue pour recherche/suivi de lésions suspectes :
des carcinomes épidermoïdes cutanés (cSCC),
des carcinomes basocellulaires cutanés (BCC),
des kératoses actiniques et des kérato-acanthomes (KA).
Des biopsies cutanées de toutes les lésions suspectes au screening ou pendant
l’étude devront être réalisées pour confirmer localement le diagnostic et traiter si
besoin selon les pratiques locales.
Carcinomes épidermoïdes non cutanés :
Examen de la tête et du cou pour vérifier l’absence de carcinomes épidermoïdes
dans la cavité buccale.
Examen anal (pour tous) et pelvien (pour les femmes) avant et en fin d’étude.
Scanner thoracique au screening, tous les 6 mois et 6 mois après arrêt du
traitement.
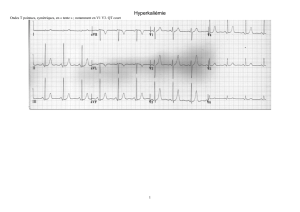
Allongement du QT :
Exclusion des patients présentant une kaliémie inférieure à la normale.
Dans le cas d’un QTc > 500 msec : suivi de l’intervalle QTc et des
électrolytes sériques jusqu’à retour à un QTc < 500 msec.
Dans le cas ou le QTc est supérieur de 60 msec par rapport au QTc
baseline, l’investigateur doit vérifier les électrolytes (K
+
, Mg²
+
et Ca²
+
) en
mettant l’accent sur l’hypokaliémie, corriger toute anomalie dans les
électrolytes avant la réintroduction du traitement, revérifier les
traitements concomitants (pour être sûr qu’aucun n’est impliqué dans la
prolongation du QTc) et éliminer ou contrôler les autres facteurs de
risques cardiaques. (i.e. : ischémie).
En cas d’arrêt temporaire du traitement, un ECG devra être réalisé le premier
jour de la réintroduction du traitement, tous les 28 jours pendant 5 cycles
(chaque J1) et tous les 3 cycles comme prévu par le protocole.
10. Questionnaire patient
Le carnet patient est un carnet d’exception : le patient doit reporter uniquement
les prises non effectuées et l’explication reliée (ex : oubli,…).
Le patient rapporte son carnet à chaque visite et repart avec ce même carnet.
11. Suivi des patients
Une visite de suivi de la tolérance sera réalisée 28 jours (+/- 5 jours) après la
dernière prise du vemurafenib (en cas de sortie prématurée ou de fin d’étude).
Les patients qui arrêtent le vemurafenib pour quelle que raison que ce soit autre
qu’un retrait de consentement, seront interrogés tous les 3 mois après la
dernière prise de vemurafenib pour évaluer la survie et recueillir les traitements
anticancéreux reçus.
Ce suivi continuera jusqu’au décès du patient, pendant une période de 12 mois
maximum après la dernière prise du traitement à l’essai.
12. Modification du traitement / Adaptations de doses
Une escalade de dose est déconseillée suite à une réduction de dose pour un
problème de tolérance : contacter Roche dans ce cas.
Aucune réduction de dose n’est prévue en cas de cancer de la peau.
Si le traitement est interrompu plus de 4 semaines du fait d’un événement
indésirable, le patient sort d’étude.
Cas particulier (à voir au cas par cas avec le promoteur) : arrêt du traitement
plus de 4 semaines pour chirurgie tumorale, radiothérapie ou autres procédures
pour des raisons de tolérance ou dans l’intérêt du patient : dans ces cas
particuliers, le vemurafenib devra être arrêté avant ces procédures et pourra être
réinitié après discussion avec Roche, sans diminution de dose.
 6
7
8
9
6
7
8
9
1
/
9
100%