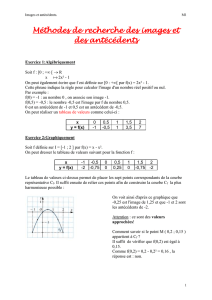
Résumé protocole BRIM 8

Résumé protocole BRIM 8
Titre de l’étude
Etude de phase III randomisée en double aveugle versus
placebo du traitement adjuvant par vemurafenib
(RO5185426) chez des patients atteints d’un mélanome
cutané avec la mutation BRAF réséqué chirurgicalement
et à grand risque de récidive.
Objectifs de l’étude
Objectif principal
L’objectif principal de cette étude est le suivant :
Évaluer l’efficacité par la survie sans maladie du
traitement adjuvant par vemurafenib.
Objectifs secondaires
Évaluer l’efficacité du traitement adjuvant par
vemurafenib mesurée par la survie globale (SG)
Évaluer l’efficacité du traitement adjuvant par
vemurafenib mesurée par la survie sans
métastase à distance (SSMD)
Évaluer l’innocuité et la tolérance du
vemurafenib en tant que traitement adjuvant
Schéma et plan de l’étude
Etude de phase III, internationale, multicentrique, en
double aveugle, randomisée, contrôlée contre placebo,
chez des patients atteints de mélanome à mutation
BRAFV600, entièrement réséqué (mutation détectée par
le test de mutation cobas® BRAF V600), présentant un
risque élevé de récidive. .
Les patients admissibles seront randomisés (1/1) pour
recevoir le placebo ou le vemurafenib pendant une
période de 52 semaines, la randomisation étant stratifiée
par stade de la maladie et par région.
Bras A : placebo par voie orale, deux fois par jour
Bras B : vemurafenib 960 mg par voie orale, deux
fois par jour

Principaux critères
d’inclusion et d’exclusion
Critères d’inclusion :
Sujets de sexe masculin ou féminin âgés de ≥ 18
ans
Patients atteints de mélanome cutané porteurs
de la mutation BRAFV600
Le patient ne doit plus présenter de maladie,
éliminée par voie chirurgicale, dans les 70 jours
précédant la randomisation
Espérance de vie d’au moins 5 ans
Les patients doivent être en rémission complète
d’éventuels effets d’une intervention chirurgicale
lourde (notamment lymphadénectomie
régionale) ou de toute lésion traumatique
significative avant l’administration de la première
dose du traitement à l’étude.
Fonctions hématologique, hépatique et rénale
adéquates, définies par les résultats biologiques
suivants, obtenus dans les 14 jours précédant la
randomisation
Les patientes en âge de procréer et les patients
ayant des partenaires en âge de procréer doivent
accepter de toujours utiliser une ou des
méthodes de contraception efficaces à compter
de la date de signature du consentement éclairé
et jusqu’à au moins 6 mois après la fin du
traitement à l’étude
Critères d’exclusion :
Antécédents de tout traitement systémique (c.-à-
d. chimiothérapie, traitement biologique ou ciblé,
ou hormonothérapie)
Antécédents de traitement par perfusion de
membre
Antécédents de radiothérapie pour le traitement
d’un cancer de la prostate, du col de l’utérus ou
du rectum

Antécédents de radiothérapie pour le traitement
du mélanome, notamment, entre autres,
radiothérapie sur un bassin ganglionnaire
réséqué
Antécédents ou preuves cliniques,
radiographiques ou pathologiques actuelles
d’atteinte ganglionnaire lymphatique récidivante
après résection d’un mélanome primaire avec
atteinte ganglionnaire lymphatique antérieure
Allergie ou hypersensibilité à des composants de
la formulation du vemurafenib
Antécédents ou preuves cliniques,
radiographiques ou pathologiques actuelles de
métastases en transit ou de lésions satellites
Antécédents de troubles cardiaques ou
pulmonaires cliniquement significatifs
Antécédents de pathologie hépatique
cliniquement significative
Infection active ou infection chronique exigeant
la prise d’antibiotiques suppressifs chroniques
Maladie auto-immune active
Autre affection médicale grave concomitante qui,
de l’avis de l’investigateur, compromettrait la
sécurité du patient ou sa capacité à participer à
l’étude
Antécédents de malabsorption ou autre trouble
métabolique cliniquement significatif
1
/
3
100%