Troubles du rythme et de la conduction chez l`enfant

Troubles du rythme et de la conduction
chez l’enfant
E. Villain
La connaissance et le traitement des troubles du rythme de l’enfant ont progressé grâce aux progrès de la
pharmacologie, de la génétique, et des méthodes interventionnelles. Les différentes tachycardies non
sinusales sont mieux connues et leur traitement codifié selon l’âge de l’enfant et l’origine de la
tachycardie. Une meilleure connaissance des traitements médicaux permet de traiter les patients avec
plus d’efficacité et de sécurité, et les troubles du rythme chroniques ou menaçants bénéficient maintenant
des méthodes ablatives ainsi que des progrès concernant le matériel de défibrillation automatique. Les
progrès de la génétique moléculaire ont bouleversé la compréhension des troubles du rythme ventriculaire
et des myocardiopathies. Enfin, le diagnostic précoce des troubles de la conduction auriculoventriculaire
permet de prendre en charge les enfants avant la naissance, et d’éviter les conséquences d’une
bradycardie sévère, puisqu’on peut désormais implanter un stimulateur à tout âge, en choisissant un
mode de stimulation adapté à la morphologie et à la pathologie de l’enfant. Nous nous limiterons
volontairement aux arythmies sur cœur sain car les séquelles rythmiques de la chirurgie cardiaque sont
très complexes et s’intègrent dans un contexte malformatif nécessitant une prise en charge très
spécialisée.
© 2006 Elsevier SAS. Tous droits réservés.
Mots clés : Tachycardie supraventriculaire ; Tachycardie atriale ; Tachycardie ventriculaire ; QT long ;
Bloc auriculoventriculaire ; Pacemaker
Plan
¶Introduction : rythme normal de l’enfant 1
¶Mécanismes des troubles du rythme cardiaque 1
Automatisme cardiaque anormal 1
Phénomènes de réentrée 2
Activités déclenchées 2
¶Tachycardies supraventriculaires (TSV) 2
Tachycardies jonctionnelles par réentrée 2
Troubles du rythme atrial 4
¶Troubles du rythme ventriculaire 6
Tachycardies ventriculaires monomorphes 6
Troubles du rythme ventriculaires et mort subite : « canalopathies » 6
¶Bradycardies de l’enfant 8
Malaises vagaux et syncopes vasovagales 8
Troubles de la conduction auriculoventriculaire 9
¶Conclusion 11
■Introduction : rythme normal
de l’enfant
L’interprétation d’un trouble du rythme chez l’enfant doit
tenir compte des données concernant le rythme normal. La
fréquence sinusale s’accélère de la naissance jusqu’à la fin du
premier mois, puis décroît ensuite après le troisième mois ; près
de la moitié de cette décroissance est obtenue avant l’âge de
2 ans.
[1-3]
Chez le nouveau-né, la fréquence maximale est
d’environ 170 à 190 battements par minute (bpm) et la fré-
quence minimale ne descend pas au-dessous de 80 bpm. Chez
le nourrisson, la fréquence maximale lors des cris peut atteindre
220, voire 240 bpm et la fréquence minimale reste en principe
supérieure à 60 bpm ; l’écart entre le jour et la nuit apparaît à
partir du troisième mois. Chez l’enfant plus grand, le ralentis-
sement est d’environ 10 à 15 bpm tous les 2 ans jusqu’à 10 ans,
puis devient moindre jusqu’à l’adolescence. La limite supérieure
du rythme sinusal à l’activité est de 180-200 bpm et les fré-
quences minimales peuvent s’abaisser jusqu’à 45 bpm ; les
pauses sinusales nocturnes n’excèdent pas 1 800 ms.
La constatation d’extrasystoles, atriales ou ventriculaires, est
rare chez le nouveau-né et le nourrisson ; elles sont habituelle-
ment bénignes et régressent dans les premiers mois. Chez
l’enfant, on observe parfois des extrasystoles ventriculaires qui
sont considérées comme bénignes lorsqu’elles sont toutes
identiques, isolées, et qu’elles disparaissent à l’effort.
Les troubles de la conduction auriculoventriculaires sont
exceptionnels chez le nourrisson normal ; chez le grand enfant,
on peut observer, sur les tracés nocturnes, des aspects de PR
long, voire des ondes P bloquées.
■Mécanismes des troubles
du rythme cardiaque
Les troubles du rythme cardiaque sont la conséquence d’une
anomalie dans la genèse de l’influx ou/et de troubles de la
conduction.
Automatisme cardiaque anormal
L’automatisme cardiaque anormal est caractérisé par la
survenue d’une dépolarisation diastolique spontanée, aboutis-
sant à un potentiel d’action dans des fibres cardiaques norma-
lement dépourvues d’automatisme. Les tachycardies par
¶4-071-A-70
1Pédiatrie
© Elsevier Masson SAS. Tous droits réservés. - Document téléchargé le 24/01/2015 par Conseil Scientifique (040888)

automatisme anormal ont pour caractéristique de ne pouvoir
être ni déclenchées ni arrêtées par stimulation cardiaque. Les
tachysystolies atriales (TSA), les tachycardies hissiennes congé-
nitales et certaines tachycardies ventriculaires (TV) relèvent de
ce mécanisme.
Phénomènes de réentrée (Fig. 1)
Le phénomène de réentrée le long d’un circuit implique la
présence de deux voies de conduction possibles pour l’influx
électrique. Un bloc unidirectionnel sur une des voies est
l’élément initiateur de la réentrée ; l’influx progresse alors le
long de l’autre voie dans laquelle la conduction doit être
suffisamment lente pour qu’il puisse ensuite emprunter dans le
sens rétrograde la première voie de conduction, devenue
perméable ; le circuit est ainsi amorcé et l’influx circule de façon
circulaire entre les deux voies de conduction. Les tachycardies
jonctionnelles réciproques et le flutter atrial obéissent à ce
mécanisme.
Activités déclenchées (Fig. 1)
Les activités déclenchées sont toujours précédées d’une
activité électrique préalable. Il s’agit de dépolarisations anorma-
les, se présentant sous la forme d’oscillations faisant suite à un
potentiel d’action, et appelées par référence à ce dernier
postdépolarisations précoces ou tardives. Ces oscillations
peuvent atteindre un potentiel seuil, initiant la dépolarisation
de la cellule, et déclencher ainsi une réponse propagée.
■Tachycardies supraventriculaires
(TSV)
Les TSV sont les troubles du rythme pédiatriques les plus
fréquents, et s’observent surtout au cours des premiers mois de
vie. Même très rapides, supérieures à 300 bpm, ces tachycardies
peuvent bénéficier d’une bonne tolérance initiale, et le diagnos-
tic repose alors sur l’auscultation. Ultérieurement, la persistance
de la tachycardie a pour conséquence l’épuisement ischémique
du ventricule gauche et une défaillance cardiaque plus ou moins
sévère. Chez le grand enfant, les accès de tachycardie sont
moins rapides, et se révèlent par des palpitations, des douleurs
thoraciques, plus rarement par des malaises, voire d’authenti-
ques syncopes.
Par définition, les TSV naissent au-dessus de la bifurcation du
faisceau de His et se caractérisent donc par des complexes QRS
fins, inférieurs à 80 ms. Le diagnostic de l’origine de la tachy-
cardie repose sur l’identification des auriculogrammes non
sinusaux P’ et sur l’analyse de la liaison entre les ondes P’ et les
complexes ventriculaires QRS (Fig. 2). Lorsque l’électrocardio-
gramme (ECG) montre autant d’ondes P’ que de QRS, ou quand
on ne voit pas bien les ondes P’, il est nécessaire de recourir à
des manœuvres vagomimétiques, qui créent un bloc auriculo-
ventriculaire (BAV) transitoire. Chez le nouveau-né, la méthode
non invasive la plus efficace pour stimuler le tonus vagal est
l’application d’une vessie de glace sur le visage durant
10 secondes ; chez l’enfant, on a recours au réflexe oculocardia-
que. En milieu hospitalier, le test à l’adénosine triphosphate
(ATP) (Striadyne
®
) est le plus utilisé pour identifier l’origine des
TSV ; l’injection se fait par voie intraveineuse flash, à la dose de
0,5 à 2 mg/kg, renouvelable, l’estomac ayant été vidé. Il est
préférable de préparer une seringue d’atropine, à injecter en cas
de pause prolongée, et l’utilisation d’ATP est déconseillée en cas
de bronchospasme.
Tachycardies jonctionnelles par réentrée
Les tachycardies jonctionnelles par rythme réciproque sont
les plus fréquentes des TSV de l’enfant, dont elles représentent
environ 70 % des cas.
[4, 5]
Leur pronostic dépend de l’âge de
l’enfant et de la nature du circuit de réentrée. Si la guérison
spontanée peut être espérée chez la plupart des enfants dont le
trouble du rythme débute dans la première année de vie, les
enfants plus grands doivent souvent être traités de façon
prolongée et certains troubles du rythme réfractaires au traite-
ment médical, persistants et/ou potentiellement dangereux, font
maintenant porter une indication d’ablation. Dans tous les cas,
une échographie doit être réalisée, une anomalie cardiaque
étant présente dans 10 à 15 % des cas.
Réentrée sur voie accessoire courante
Mécanisme
Durant les accès de tachycardie, la voie normale nodohis-
sienne est empruntée des oreillettes vers les ventricules, et la
12
B
C
D
Figure 1. Mécanismes des troubles du rythme.
A. Mécanisme de réentrée. L’influx bloque sur une des voies de conduc-
tion (grisé) qui est empruntée ensuite de façon rétrograde, amorçant ainsi
le circuit. 1. Réentrée jonctionnelle sur voie accessoire : l’influx descend
par la voie normale et remonte par la voie accessoire atrioventriculaire ; 2.
réentrée intranodale.
B. Syndrome de Wolff-Parkinson-White. 1. En rythme sinusal, l’influx
emprunte à la fois la voie nodohissienne (frein vagal) et la voie accessoire ;
2. fibrillation atriale conduite simultanément par la voie normale, et par la
voie accessoire.
C. Postdépolarisations tardives.
D. Postdépolarisations précoces.
Identification des auriculogrammes P'
(spontanés ou manœuvres vagales)
P > QRS
Tachycardie atriale
- flutter
- tachysystolie
- tachycardie atriale chaotique
P = QRS
Réentrée jonctionnelle
- voie accessoire
- réentrée intranodale
P < QRS (ou dissociation)
Tachycardie hissienne
Figure 2. Diagnostic de l’origine d’une tachycardie supraventriculaire.
4-071-A-70
¶
Troubles du rythme et de la conduction chez l’enfant
2Pédiatrie
© Elsevier Masson SAS. Tous droits réservés. - Document téléchargé le 24/01/2015 par Conseil Scientifique (040888)

voie accessoire (faisceau de Kent) est empruntée dans le sens
rétrograde (Fig. 1A). L’ECG montre donc une tachycardie à QRS
fins, entre 250 et 320 bpm, comportant autant de complexes
QRS que d’ondes P’ rétrogrades, puisque oreillettes et ventricules
sont activés à chaque tour du circuit (Fig. 3A). Les ondes P’ sont
habituellement bien visibles après les complexes QRS et leur axe
peut même permettre de déterminer le siège de l’émergence de
la voie accessoire. Les manœuvres vagales entraînent l’arrêt
brutal de la crise dans 80 % des cas, en bloquant le circuit de
réentrée dans le nœud auriculoventriculaire ; si le circuit n’est
pas interrompu, la tachycardie n’est pas modifiée : c’est la loi du
tout ou rien. La cardioversion (1-3 J/kg chez un nourrisson) n’a
d’intérêt qu’en cas d’échec des manœuvres vagales, pour arrêter
une réentrée menaçant les fonctions vitales. L’injection intra-
veineuse d’antiarythmiques est dangereuse chez les nourrissons,
avec un risque de collapsus cardiovasculaire ou de proarythmie
irréversible.
L’étude du tracé intercritique montre parfois un aspect de
préexcitation ventriculaire, définissant un syndrome de Wolff-
Parkinson-White (WPW). En rythme sinusal normal, l’influx
électrique circule simultanément dans la voie nodohissienne et
dans la voie accessoire (dépourvue du frein vagal), si bien qu’un
des ventricules est activé avant l’autre : il en résulte un aspect
de préexcitation avec PR court et onde delta sur l’ECG de
surface (Fig. 4).
Syndrome de Wolff-Parkinson-White et perméabilité
antérograde de la voie accessoire
Toute la gravité du syndrome de WPW tient au risque de
syncope, voire de mort subite à laquelle sont exposés les
patients. Ces accidents sont la conséquence de la conduction
très rapide vers les ventricules d’un trouble du rythme atrial
(fibrillation ou flutter) à travers une voie accessoire très
perméable, court-circuitant le nœud auriculoventriculaire
(Fig. 1). Habituellement, ce trouble du rythme survient à
l’activité, et la plupart des cas de mort subite chez les enfants
concernaient de jeunes sportifs.
[6, 7]
Ces accidents sont d’autant
plus rares que l’enfant est plus jeune, et leur fréquence est
d’autant plus difficile à estimer que les études d’évolution
spontanée sont rares.
[8, 9]
Lors d’une fibrillation atriale conduite par une voie acces-
soire, l’ECG montre une tachycardie irrégulière, avec des QRS
plus ou moins larges (plus ou moins « préexcités ») car l’influx
en provenance des oreillettes est conduit à la fois par la voie
normale et par la voie accessoire (Fig. 4). Le traitement de ce
trouble du rythme varie selon la tolérance clinique. Les formes
avec collapsus doivent être traitées par choc électrique externe.
Lorsque le trouble du rythme est bien supporté, il peut être
réduit par l’injection intraveineuse de flécaïnide ou d’amioda-
rone. L’utilisation d’agents déprimant la conduction (Stria-
dyne
®
) est contre-indiquée, car ces drogues favorisent la
transmission des influx atriaux par la voie accessoire et indui-
sent donc une accélération de la fréquence ventriculaire. C’est
également en raison de ce risque que la digoxine est contre-
indiquée chez les patients ayant un syndrome de WPW.
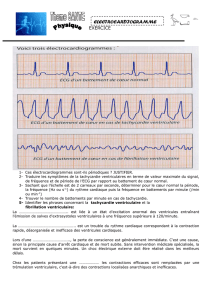
Figure 3. Tachycardie supraventriculaire (TSV) du nouveau-né et du
nourrisson.
A. TSV sur voie accessoire courante : tachycardie à QRS fins, à 280 bpm,
avec des ondes P’ derrière les QRS (P’ = QRS).
B. Tachycardie jonctionnelle réciproque permanente. Tachycardie à
220 bpm avec QRS-P’ long et ondes P’ rétrogrades négatives en DII, DIII et
aVF.
C. Test à l’adénosine triphosphate (ATP) (Striadyne
®
) ; en haut : l’injection
d’ATP « casse » la tachycardie avec retour en rythme sinusal. Il s’agit donc
d’une TSV par réentrée jonctionnelle ; en bas : l’injection d’ATP crée un
bloc auriculoventriculaire (BAV) transitoire, qui laisse apparaître les ondes
P’ d’une tachycardie atriale ectopique (P’ négatives en DII).
Figure 4. Syndrome de Wolff-Parkinson-
White. En haut : électrocardiogramme en
rythme sinusal, avec PR court et onde delta sur
les ventriculogrammes. En bas : fibrillation
atriale conduite par la voie accessoire. Les QRS
ont une morphologie variable et l’espace RR le
plus court correspond à une fréquence ventri-
culaire > 300 bpm.
Troubles du rythme et de la conduction chez l’enfant
¶
4-071-A-70
3Pédiatrie
© Elsevier Masson SAS. Tous droits réservés. - Document téléchargé le 24/01/2015 par Conseil Scientifique (040888)

S’il existe une majorité de formes bénignes, des cas de
fibrillation ventriculaire ou de mort subite ont été rapportés
chez des enfants asymptomatiques, n’ayant jamais fait d’accès
de TSV, et aucune méthode ne permet avec certitude de prédire
quels sont les patients à risque. Le caractère intermittent d’une
préexcitation et la disparition brutale de l’onde delta à l’épreuve
d’effort sont en faveur d’une voie accessoire peu perméable dans
le sens antérograde. L’étude électrophysiologique endocavitaire
est considérée comme le moyen le plus fiable pour détecter les
formes malignes de syndrome de WPW, définies habituellement
par la présence de deux critères :
• conduction rapide dans le faisceau de Kent lors de la stimu-
lation atriale ;
• déclenchement d’une fibrillation atriale soutenue avec RR
court.
[7, 10]
.
L’évaluation endocavitaire peut être remplacée par une étude
par voie transœsophagienne,
[11, 12]
que nous réalisons vers l’âge
de8à10ans. Une étude randomisée récente a montré un
risque d’accidents graves chez des enfants non traités, ayant un
syndrome de WPW considéré comme dangereux,
[10]
et une
ablation prophylactique de la voie accessoire est donc conseillée
chez ces patients.
Traitement des TSV sur voie accessoire du nouveau-né
et du nourrisson
Les récidives de TSV par réentrée sur voie accessoire sont
fréquentes dans les premiers mois de vie et on traite donc
médicalement les nourrissons âgés de moins de 1 an. Classique-
ment, ce traitement reposait sur la digoxine, même en cas de
préexcitation ventriculaire, car on considérait que le risque de
fibrillation ventriculaire était quasi nul à cet âge. Cependant, un
travail concernant 141 nourrissons âgés de moins de 1 an a
montré que trois nouveau-nés, dont deux avaient un syndrome
de WPW, avaient eu une fibrillation ventriculaire en début de
traitement par la digoxine ; en outre, la digoxine n’avait été
efficace que dans 65 % des cas pour prévenir les récidives de
TSV.
[13]
L’amiodarone orale s’était en revanche révélée très
efficace (97 % de succès) avec des effets secondaires rares sous
couvert d’une surveillance thyroïdienne. Suite à cette étude et
à d’autres publications faisant état de cas de fibrillation
ventriculaire sous digoxine, chez des nourrissons ayant un
syndrome de WPW, la prévention des récidives des TSV du
nourrisson dans notre département est désormais assurée par
l’amiodarone ; en cas d’échec, on y associe un bêtabloquant. Le
traitement doit être maintenu environ 1 an et la plupart des
enfants ne font pas de récidive lors du sevrage. Une surveillance
régulière est poursuivie en cas de préexcitation patente sur
l’ECG de surface, d’autant qu’on a montré chez ces patients un
risque de récidive tardive de TSV par réentrée, à un âge moyen
de 8 ans.
[14]
Traitement des TSV sur voie accessoire du grand enfant
Lorsque les premières crises de TSV surviennent dans l’en-
fance, la guérison spontanée est rare et la décision de donner
un traitement médical préventif journalier dépend alors de la
fréquence des crises, des facteurs déclenchants éventuels (stress,
effort) et de la tolérance clinique. En l’absence de préexcitation
ventriculaire, tous les antiarythmiques méritent d’être essayés.
Sinon, notre préférence va à des bêtabloquants de longue durée
d’action (sotalol ou nadolol) en cas de crises liées à l’effort ou
bien à des antiarythmiques de la classe IC (flécaïnide, propafé-
none) qui allongent la période réfractaire des voies accessoires ;
l’amiodarone est réservée aux formes réfractaires au traitement
par ces agents.
Les méthodes ablatives permettent maintenant de guérir
définitivement les enfants ayant une TSV chronique ou réfrac-
taire au traitement médical, ceux ayant fait une syncope, ainsi
que ceux ayant un syndrome de WPW considéré comme
dangereux. Cette méthode consiste à détruire les structures
myocardiques responsables des troubles du rythme, voie
accessoire en l’occurrence, grâce à l’application d’un courant
électrique (radiofréquence) ou de froid (cryothérapie). Ce
procédé est très efficace, avec des taux de succès supérieurs à
90 % chez les enfants, si on s’adresse à des équipes
expérimentées.
[15]
Les complications sont rares et s’observent surtout dans
certaines localisations ; ainsi, l’ablation des voies accessoires
septales peut se compliquer de BAV et des complications
coronariennes ont été décrites après ablation de voies accessoi-
res droites. Pour éviter la constitution de lésions définitives des
voies de conduction normales, on peut utiliser la cryothérapie
plutôt que l’application d’un courant de radiofréquence.
[16]
Tachycardie jonctionnelle réciproque permanente
Les tachycardies jonctionnelles réciproques permanentes
représentent moins de 10 % des TSV de l’enfant, mais il s’agit
d’un trouble du rythme incessant, chronique, et difficile à
traiter. Le circuit de réentrée s’établit entre la voie nodohis-
sienne normale, empruntée dans le sens antérograde, et une
voie accessoire qui n’est perméable que dans le sens rétrograde,
à conduction lente et qui émerge dans l’oreillette à proximité
du sinus coronaire. Ce trouble du rythme est facile à diagnosti-
quer sur l’ECG de surface, qui montre des ondes P’ rétrogrades
éloignées du complexe QRS précédent (QRS-P’ long) et négatives
dans les dérivations inférieures DII, DIII et aVF (Fig. 3B). Les
manœuvres vagales interrompent transitoirement le circuit ou
bien n’ont aucun effet, ce qui permet de différencier cette
réentrée d’un foyer ectopique atrial caudal. Ce trouble du
rythme est souvent peu rapide, de l’ordre de 200 bpm, et peut
alors passer inaperçu, entraînant progressivement une
défaillance cardiaque sévère ; parfois, la TSV évolue par accès
paroxystiques, d’où la règle de faire systématiquement un
enregistrement ECG des 24 heures devant une myocardiopathie
inexpliquée de l’enfant.
Le traitement médical des tachycardies jonctionnelles réci-
proques permanentes repose sur l’amiodarone chez les jeunes
enfants, puis sur les bêtabloquants, les calcium-bloquants et la
propafénone chez l’enfant plus grand. Le pourcentage de
guérison spontanée est d’environ 30 % des patients, mais il faut
souvent attendre jusqu’à l’adolescence pour pouvoir arrêter le
traitement. Comme l’ablation donne d’excellents résultats, elle
n’est plus seulement réservée aux formes mal tolérées, réfractai-
res au traitement ou qui ne disparaissent pas avec l’âge ; on
peut également la proposer vers l’âge de8à10ansdans les cas
d’enfants et de familles ne souhaitant pas poursuivre un
traitement médical prolongé.
[17, 18]
Rythmes réciproques par réentrée intranodale
Après l’âge de 10 ans, les réentrées intranodales constituent
la cause la plus fréquente de TSV.
[19]
Le terme de maladie de
Bouveret est souvent utilisé en France pour désigner ce trouble
du rythme, dont le circuit fait intervenir deux voies de conduc-
tion développées dans le nœud auriculoventriculaire (Fig. 1). Il
s’agit d’une TSV régulière, souvent peu rapide (≤200 bpm) avec
des ondes P’ rétrogrades qu’on ne voit pas, ou bien qui défor-
ment la fin des complexes QRS ; en effet, le temps que met
l’influx pour descendre vers les ventricules est sensiblement le
même que celui qu’il met pour remonter la voie rapide vers les
oreillettes. Les accès de TSV intranodales sont en règle bien
tolérés et ne méritent aucun traitement de fond. En cas de crises
répétées, on a le choix entre un traitement médical ou bien une
ablation, maintenant choisie par un nombre croissant de
patients, gênés par le handicap que représentent des crises
fréquentes et imprévisibles.
[20]
Troubles du rythme atrial
Le diagnostic de l’origine atriale d’une TSV se fait sur l’ECG
de surface, qui montre plus d’ondes P’ que de QRS, soit spon-
tanément, soit à la faveur d’une manœuvre vagale (Fig. 3).
Flutter atrial néonatal
Le flutter est un trouble du rythme fréquent, survenant chez
des nouveau-nés ayant un cœur normal, caractérisé par des
ondes P en « dents de scie » sans retour à la ligne isoélectrique.
4-071-A-70
¶
Troubles du rythme et de la conduction chez l’enfant
4Pédiatrie
© Elsevier Masson SAS. Tous droits réservés. - Document téléchargé le 24/01/2015 par Conseil Scientifique (040888)

La fréquence atriale est très rapide, environ 400 bpm et la
conduction vers les ventricules se fait habituellement en 2 : 1,
si bien que la tolérance clinique est initialement bonne. Le
flutter est une réentrée, dont la réduction peut être obtenue par
stimulation atriale rapide. Comme la proximité anatomique de
l’oreillette gauche et de l’œsophage permet de stimuler
l’oreillette par voie transœsophagienne, cette méthode est
devenue le mode électif de réduction du flutter néonatal
(Fig. 5). En cas d’échec, la stimulation endocavitaire est presque
constamment efficace. Si on ne dispose pas de matériel de
stimulation, la digoxine seule ou associée à l’amiodarone orale
est efficace, mais d’action lente ; en cas de détresse circulatoire
liée à la conduction rapide d’un flutter vers les ventricules, il
faut donc faire un choc électrique. Après retour en rythme
sinusal, le traitement d’entretien est la digoxine durant 3 mois
et les enfants sont définitivement guéris.
[21, 22]
Tachysystolies atriales ou tachycardies atriales
monomorphes
Les TSA sont dues à l’automaticité anormale d’un foyer
ectopique auriculaire. Dans les cas simples, il y a plus d’ondes
P’ que de QRS et leur morphologie est différente de l’onde P
sinusale. Le diagnostic est plus difficile à faire en cas de foyer
situé dans la partie haute de l’oreillette droite ou à proximité de
la veine pulmonaire supérieure droite, car l’auriculogramme a
alors la morphologie et l’axe d’une onde P normale, et ces TSA
ressemblent donc à des tachycardies sinusales. Dans ces condi-
tions, le diagnostic différentiel entre une TSA responsable d’une
myocardiopathie et une tachycardie sinusale secondaire à une
myocardiopathie primitive peut être difficile à faire.
[23, 24]
L’attention doit être attirée par un espace PR un peu long, et les
manœuvres vagales sont très utiles pour démasquer les ondes P’
en créant un BAV transitoire (Fig. 3) ; l’enregistrement Holter
montre parfois des ondes P bloquées, indiquant l’origine
ectopique du rythme atrial.
Ces tachycardies sont difficiles à traiter. Les bêtabloquants,
seuls ou associés à l’amiodarone et/ou à la digoxine, sont les
drogues les plus efficaces, de même que les agents de la classe
IC. À long terme, le pronostic est largement fonction de l’âge
de l’enfant. Quand la TSA débute avant l’âge de 3 ans, le
pronostic est en règle excellent, car la plupart de ces foyers
ectopiques disparaissent spontanément avec la croissance.
Quand le trouble du rythme débute plus tardivement, la TSA
récidive fréquemment lors de l’arrêt du traitement et seule
l’ablation par radiofréquence permet de restaurer un rythme
sinusal stable.
[23-26]
Tachycardie atriale chaotique
Cette tachycardie s’observe exclusivement chez les nourris-
sons et son diagnostic est très facile en raison du polymor-
phisme de l’activation atriale. Le trouble du rythme est souvent
dépisté lors de l’auscultation, du fait de l’irrégularité du rythme
cardiaque, mais les formes rapides peuvent entraîner une
défaillance cardiaque. Sur l’ECG, on voit de multiples foyers
d’activation atriale (au moins trois morphologies de P), avec des
aspects de bloc de branche et des ondes P bloquées ; des
passages en flutter et en fibrillation sont souvent visibles
(Fig. 6). Les formes rapides sont traitées par la digoxine, le
sotalol ou l’amiodarone et la guérison définitive est la règle
après 12 à 18 mois de traitement.
[27]
Tachycardies hissiennes congénitales
Les tachycardies hissiennes sont les plus rares des TSV du
nourrisson mais les plus graves, car elles peuvent évoluer vers
la défaillance cardiaque et la mort, en l’absence de traitement.
Ces tachycardies sont liées à l’activité d’un foyer ectopique situé
dans le faisceau de His, dont la fréquence est le plus souvent
comprise entre 150 et 280 bpm. Dans les formes typiques, l’ECG
montre une tachycardie à QRS fins avec dissociation auriculo-
ventriculaire et complexes de capture (Fig. 7). Dans les cas avec
conduction rétrograde ventriculoatriale 1 : 1, l’injection de
Striadyne
®
conduit au diagnostic en créant un bloc rétrograde
(QRS > P’). Les chocs électriques n’ont habituellement pas
d’effet, car le foyer automatique reprend immédiatement son
activité. L’amiodarone est le traitement le plus efficace des
tachycardies hissiennes ; habituellement, le traitement ne réduit
pas le trouble du rythme mais ralentit la fréquence du foyer
d’automatisme, en attendant que le sinus reprenne la com-
mande de l’activité électrique du cœur, parfois après plusieurs
années, ce qui permet alors le sevrage thérapeutique.
[28]
D’autres agents tels que la flécaïnide et la propafénone sont
efficaces mais d’un maniement plus difficile chez le jeune
enfant.
Figure 5. Réduction d’un flutter néonatal par stimulation œsopha-
gienne.
A. Flutter atrial, bloqué 2/1 vers les ventricules.
B. Ondes de flutter démasquées lors de l’injection d’adénosine triphos-
phate (ATP).
C. Stimulation atriale en « rafale », suivie du retour en rythme sinusal.
Figure 6. Tachycardie atriale chaotique. L’activité atriale est irrégulière,
polymorphe et instable. On voit également des ondes P bloquées et des
aspects intermittents de bloc de branche fonctionnel.
Figure 7. Tachycardie hissienne. La fréquence ventriculaire est à
200 bpm, et on voit une activité sinusale (P) dissociée et plus lente.
Troubles du rythme et de la conduction chez l’enfant
¶
4-071-A-70
5Pédiatrie
© Elsevier Masson SAS. Tous droits réservés. - Document téléchargé le 24/01/2015 par Conseil Scientifique (040888)
 6
7
8
9
10
11
12
13
6
7
8
9
10
11
12
13
1
/
13
100%