CHI 110 - STRUCTURE DE LA MATIERE 3ème PARTIE : LES

CHI 110 - STRUCTURE DE LA MATIERE
NOTES DE COURS
3ème PARTIE : LES ETATS DE LA MATIERE
I. Généralités
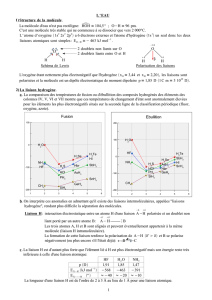
Les trois états de la matière : gazeux, liquide, solide
Ces trois états se distinguent par l’importance des forces de cohésions qui existent entres les
composants (éléments, ions, molécules) de la matière.
Attention : la matière peut changer d’état selon la température et la pression (exemple de
l’eau).
II – 1. Gaz
Un gaz est composé de molécules, ou d’atomes (cas des gaz rares), qui sont en très faible
interaction. On peut considérer sue les espèces sont pratiquement indépendantes.
II – 1. Liquides
L’état liquide est un état intermédiaire entre gaz et solide. Il existe entre les molécules des
forces de cohésion qui limitent leur mouvement.
II – 1. Solides
L’état solide est un état à la fois condensé et ordonné : il n’y a plus de mouvement des
différentes espèces qui composent le solide. Il existe différents types de solides :
- Les solides ioniques (ex. : NaCl) : les liaisons entre les éléments qui les composent
sont fortes (167-630 kJ mol–1), mais en général ils sont facilement solubles dans l’eau.
- Les solides métalliques (ex. : Na métal) : la liaison métallique est moins forte que la
liaison ionique, ce qui se traduit en particulier par le fait que les métaux peuvent être
Liquide
état condensé
Gaz
Solide
état très condensé
(ordonné) augmentation de la
force des
interactions entre
les espèces
pas,
ou très peu,
d’interactions
des forces de cohésion
faibles (liaisons
hydrogène, liaisons de
Van der Walls) limitent le
mouvement des espèces
interactions très fortes :
solides ioniques,
métalliques et
moléculaires (liaisons
covalentes)
Liquide
état condensé
Gaz
Solide
état très condensé
(ordonné) augmentation de la
force des
interactions entre
les espèces
pas,
ou très peu,
d’interactions
des forces de cohésion
faibles (liaisons
hydrogène, liaisons de
Van der Walls) limitent le
mouvement des espèces
interactions très fortes :
solides ioniques,
métalliques et
moléculaires (liaisons
covalentes)

2
déformés sous l’action de forces extérieures (il faut éventuellement les chauffer pour
pouvoir les déformer). Propriétés particulières : conduisent la chaleur et l’électricité.
- Les solides moléculaires
Exemple : le graphite (nC, type sp2). Il est composé de plans dont la cohésion est assurée par
des liaisons faibles du type Van der Walls (voir plus loin).
Les liaisons covalentes sont fortes (et peu polarisées dans ce cas) et difficiles à rompre.
Les liaisons entre les plans sont faibles : on peut en particulier faire glisser les plans les
uns par rapport aux autres.
Dans le plan, les liaisons π sont délocalisés Ö conduction du courant dans ce plan.
CC
C
CC
C
CC
C
C
C
CC
C
C
C
C
CC
CC
CCC
C
CC
C
C
CCC
C
CC
C
CC
CC
CCC
C
CC
C
C
CCC
C
CC
C
CC
CC
CCC
C
CC
C
C
CCC
C
CC
C
CC
- Les solides covalents
Exemple : le diamant (nC). Il est composé uniquement d’atomes de carbone (type sp3), tous
reliés par des liaisons covalentes fortes. Le diamant est un solide très dur, qui ne se déforme
pas. Il est relativement chimiquement inerte, insoluble dans l’eau, …
CC
C
CC
C
C
C
C
C
C
C
C
CCCC
C
C
C
C
C

3
II. Les gaz : loi des gaz parfaits
L’état gazeux se caractérise par son aspect non condensé. L’espacement important entre les
molécules ou les atomes qui le constituent lui confère des propriétés particulières. Ces
propriétés sont relativement simples, ce qui a permis aux premiers chimistes de l’ère moderne
de découvrir des notions fondamentales sur les atomes et les molécules.
• Tous les gaz sont miscibles entre eux, car il n’y a que peu (ou pas du tout) d’interactions
entre les espèces à l’état gazeux.
• Les gaz présentent les 4 caractéristiques fondamentales suivantes :
1. leur composition : si dans un mélange de gaz ni est le nombre de moles d’un
gaz i, le nombre total de moles nT de gaz dans ce mélange sera nT = Σni ;
2. leur température, qui sera donnée en K (t en °C + 273,15) ;
3. leur volume (en m3) ;
4. leur pression (en N/m2).
• Les relations entre pression, volume, température, sont définies par trois lois :
¾ à T cste, PV = cste (loi de Boyle-Mariotte) : si P , V (transformation isotherme) ;
¾ à P cste, T
V = cste (loi de Gay-Lussac/Charles) : si T , V (transformation isobare) ;
¾ à V cst, T
P = cste (pas de nom pour cette loi) : si T , P (transformation isochore).
¾ Ces trois lois se résument en une seule :
T
PV = cste
Cette constante ne dépend que du nombre de moles de gaz nT (indice T pour total) et d’une
constante R, qui est appelée constante des gaz parfaits. C’est la loi d’Avogadro-Ampère.
T
TT
T
VP = nTR ( ou PTVT = nTRTT)
Remarques :
¾ la relation entre les propriétés macroscopiques des gaz est liée uniquement au nombre
de molécules, pas à leur masse ;

4
¾ les gaz réels ne suivent qu’approximativement cette loi simple ; la loi des gaz parfaits
n’est valable qu’à faible pression ;
¾ cette équation traduit le comportement des gaz par ses paramètres physiques (équation
d’état) ;
¾ c’est à cette occasion qu’a été introduite la notion de mole (qui signifie une grande
quantité de molécules. De façon plus exacte, cette quantité est le nombre d’Avogadro :
N = 6,022¯1023 unités par mole.
• Soit xi la fraction molaire d’un gaz i dans un mélange :
xi =
i
i
n
n
Σ =
T
i
n
n ; Σxi = 1
• Soit Pi la pression partielle du gaz i dans un mélange :
Pi = xiPT
• Autres variables :
- la concentration c = V
n ; d’après la loi des gaz parfaits c = RT
P
- la masse volumique ρ = V
m ; comme m = nM (où M est la masse molaire), ρ = RT
PM
- la densité d =
air
ρ
ρ
; comme la masse molaire moyenne de l’air est de 29g mol–1, on a
d = 29
M.
Remarques :
¾ Rappel sur les unités : température en degrés Kelvin K (t en °C + 273,15) ; volume en
m3 ; pression en N/m2 ;
¾ R (cste des gaz parfaits) = 0,082056 l.atm.K–1.mol–1 ;
¾ R (dans le SI) = 8,314 J.K–1.mol–1 ; on voit donc apparaître la dimension d’une
énergie. En effet, PV force¯volume/surface = force¯longueur : c’est un travail, donc
de l’énergie.
¾ Forme ″pratique″ de cette loi : V = P
nRT ;
Dans les conditions standard (T = 273,15 K, P = 1 atmosphère), le volume de 1 mole de gaz
vaut 22,414 litres (valable pour tous les gaz).

5
III. Les forces faibles (liaisons non covalentes)
Les molécules, formées d’atomes maintenus ensemble par des liaisons chimiques covalentes
ou covalentes polarisées, ne peuvent exister sous forme isolée que dans les gaz à très faible
pression. Dans les milieux condensés – liquides, solutions, ou solides – ainsi que dans les gaz
à des pressions ordinaires, les molécules entrent en contact avec d’autres molécules qui
peuvent être du même type (cas d’un liquide), soit différentes (cas d’une solution). Dans ces
conditions il existe un certain nombre d’interactions d’origine électrostatique qui
produisent des forces intermoléculaires, responsables de la formation de liquides ou solides
à température ordinaire, ainsi que de l’organisation de la matière (micelles, vésicules,
membranes, ….).
III – 1. Forces intermoléculaires (ou forces de Van der Walls)
Rappel : la loi des gaz parfaits (PV = nRT) n’est valable que pour les gaz à très faible
pression. Pour les gaz sous haute pression (gaz réels), cette relation devient :
()
RTbV
V
a
P=−
⎟
⎠
⎞
⎜
⎝
⎛+2
Dans cette relation empirique, établie par VAN DER WALLS, le terme correctif 2
V
a tiens
compte des forces d’attraction entre les molécules, créant ainsi des liaisons faibles, de l’ordre
de 8 à 40 kJ mol–1. Ces forces sont d’origine électrostatique et sont liées soit à la polarité
d’une molécule polaire, soit à l’aptitude d’une molécule apolaire à se polariser (polarisabilité).
Les forces intermoléculaires (ou forces de Van der Walls) correspondent à des interactions
entre molécules à courtes distances (0,3 à 0,8 nm) qui produisent une force totale d’attraction
intermoléculaire responsable de la cohésion de la matière dans les milieux condensés. A très
courtes distances (d<0,3 nm) ces interactions deviennent répulsives et empêchent les
molécules de se rassembler pour atteindre de très fortes densités (e.g. celles des noyaux). Il
existe donc une distance optimale qui correspond au maximum d’attraction entre deux
molécules
On rappelle encore une fois que l’origine de ces interactions est électrique.
Elles seront présentées ici par ordre d’énergie décroissante.
 6
7
8
9
10
11
6
7
8
9
10
11
1
/
11
100%