canal_bleu

Cas clinique
un «Canal bleu»
Caroline BONNET
C.H.U. Bocage, DIJON
Il s’agit d’une jeune-femme âgée de 29 ans d’origine cam-
bodgienne en France depuis quelques mois. Elle consulte
pour bilan de dyspnée au moindre d’effort et accès de
palpitations. Cette symptomatologie est ancienne et s’est
accentuée depuis 1 an. Elle n’a pas de facteurs de risque
cardiovasculaires ni d’antécédents notables hormis la notion
de «problème cardiaque» à la naissance classé sans suite.
Il n’y a pas d’antécédents familiaux. Elle ne prend pas de
traitement et elle n’a pas d’allergie.
A l’examen sa tension artérielle est normale. Il existe une
désaturation prédominant aux membres inférieurs à 76%
contre 88% aux membres supérieurs. Les bruits du cœur
sont réguliers avec un éclat de B2 au foyer pulmonaire. Les
pouls sont tous perçus et il n’y a pas de signes d’insuffi-
sance cardiaque.
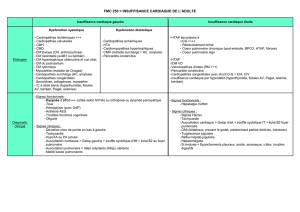
L’ECG s’inscrit en rythme sinusal avec une surcharge
droite (Image 1). A la radiographie pulmonaire, il existe
une saillie importante de l’arc moyen gauche évoquant
une dilatation de l’artère pulmonaire tronc (APT). Il n’y a
pas de surcharge vasculaire pulmonaire. Il n’y a pas de
cardiomégalie (Image 2).
A l’échocardiographie, on retrouve une dilatation importante
des cavités droites avec une hypertrophie ventriculaire
droite (Image 3). Les cavités gauches sont de tailles nor-
males mais déviées. Le septum inter ventriculaire est plat en
faveur d’une pression ventriculaire droite au moins iso sys-
témique (Image 4). En para sternal petit axe (PSPA), on
confirme les données de la radiographie pulmonaire avec
une dilatation importante de l’APT à 44mm et des branches
pulmonaires (Image 5). Il existe une hypertension artérielle
pulmonaire systolique (HTAP) estimée sur le flux IT à
105mmHg (Image 6). Il n’est pas retrouvé d’obstacle sur la
voie éjection droite. Le flux trans-valvulaire pulmonaire est
typique avec l’encoche méso systolique témoignant d’une
HTAP importante (Image 7). La fonction d’éjection ventricu-
laire droite st conservée comme en témoigne le TAPSE (20
mm) et la mesure de l’onde S à l’anneau (10.3cm/s).
Sur la base de ces premières constations, les hypothèses
diagnostiques sont une HTAP secondaire à une cardiopathie
congénitale (communication inter auriculaire associée ou
non à un retour veineux pulmonaire anormal, communica-
tion inter ventriculaire…) ou secondaire à une cardiopathie
notamment valvulaire. L’échocardiographie ne retrouve
d’anomalies architecturales ni de cardiomyopathie ou encore
de valvulopathie gauche. L’hypothèse d’une fenêtre aorto-
pulmonaire ou d’une HTAP primitive sont à évoquer mais
cela n’explique pas l’asymétrie de désaturation observée
entre les membres supérieurs et inférieurs.
Figure 1 Figure 2
Figure 3
Figure 4
Figure 5
46 • mARs 2012 - N°27
SESSION d’OCTOBRE 2011

mARs 2012 - N°27 • 47
L’incidence para sternal gauche petit axe (PSPA) gauche
permet de faire le diagnostic en montrant en 2D un large
canal artériel (Image 8). Son implantation est habituelle au
toit de l’APT. Le doppler couleur retrouve un shunt exclusi-
vement droit-gauche (Image 9). La visualisation du shunt
droit-gauche est d’autant plus difficile que les repères ana-
tomiques sont modifiés par cette dilatation de l’APT et que
l’on peut prendre ce canal abusivement pour l’artère pul-
monaire gauche.
Le cathétérisme cardiaque droit confirme les données de
l’échocardiographie. L’HTAP est systolo diastolique supra
systémique (systolique/diastolique/moyenne) à
135/77/103mmHg pour une pression aortique (systolique/
diastolique/moyenne) à 98/75/87 mmHg). Les pressions de
remplissage droit et gauche sont normales. La mesure des
saturations ne retrouve pas de shunt à l’étage auriculaire ni
ventriculaire mais un shunt à l’étage artériel avec une désa-
turation prédominant aux membres inférieurs. Le trajet de
la sonde est typique avec une image reproduisant la lettre
« phi » correspondant au passage de la sonde dans le
canal depuis l’artère pulmonaire tronc (Image 10).
Un traitement par bosentan a été débuté ainsi qu’une sup-
plémentation en fer pour lutter contre la microcytose
secondaire à la polyglobulie associée à une contraception
orale. A 6 mois, la patiente se dit moins dyspnéique comme
en témoigne le test de marche de 6 min qui a 450m versus
342m avant la mise en place du traitement par bosentan.
Les données échocardiographiques sont inchangées.
La deuxième partie de cette présentation sera consacrée à
la persistance du canal artériel en excluant volontairement
la période néonatale. Le canal artériel se développe à par-
tir de la portion distale du 6ème arc aortique gauche. En fin
de grossesse, il relie le toit de l’artère pulmonaire tronc et
l’isthme. Sa fermeture fonctionnelle a lieu dans les 24-48h
de vie. Sa fermeture définitive avec formation d’un tissu
fibreux (ligament artériel) dans un délai de 2-3 semaines.
On parle de Persistance du canal artériel (PCA) au-delà du
3ème mois de vie. Sa fréquence est de 1 pour 2500-5000
naissances soit 9-12% des cardiopathies congénitales un
sex ratio 2 à 3 filles pour un garçon.
Les modalités de découverte sont variables depuis la
période néonatale jusqu’à l’âge adulte. Schématiquement,
on distingue le CA silencieux découvert fortuitement. En
l’absence de souffle, l’abstention thérapeutique est de
mise. La 2ème situation est un canal artériel persistant
découvert sur un bilan de souffle sous clavier gauche. La
symptomatologie est fonction du diamètre et du rapport
Figure 10
Figure 6
Figure 7
Figure 8
Figure 9

48 • mARs 2012 - N°27
Figure 11
Figure 12
Figure 13
Figure 15
Figure 16 Figure 17 : Anoop et al,
NEJM 2011(17); 364: 7.
Figure 14
des résistances vasculaires pulmonaires et systémiques (RVP/RVS). En cas
de PCA modéré, on aura en échocardiographie une surcharge des cavités
gauches et une dilatation de l’APT. La 3ème situation est le cas de la jeune-
femme qui correspond à un large CA négligé ou non traité ayant évolué vers
l’HTAP. Dans ce cas, il existe une contre-indication à fermer le canal artériel
qui au contraire est une voie de décharge pour le ventricule droit. Cette voie
de décharge explique la relative «bonne tolérance» des patients ayant un
syndrome d’Eisenmenger (HTAP secondaire à une cardiopathie congéni-
tale) par rapport aux autres étiologies d’HTAP.
L’examen de référence pour faire le diagnostic de PCA est l’échocardiogra-
phie. L’incidence PSPA gauche permet de dégager la bifurcation pulmo-
naire. S’il y a un canal, on le visualisera au doppler couleur d’autant plus
facilement que le shunt est gauche-droit. Dans cette incidence, l’aligne-
ment est optimal pour mesurer le gradient systolique pic à pic entre l’aorte
descendante et l’APT (Images 11 et 12). L’incidence supra sternale permet
aussi la visualisation d’un PCA surtout chez l’enfant d’autant plus facilement
que le shunt est gauche-droit (Image 13).
Généralement les canaux artériels persistants sont accessibles à une fer-
meture per cutanée. Le recours à la chirurgie est rare en dehors de la
période néonatale. Il existe une classification des CA persistant en fonction
de l’aspect anatomique (Image 14). Schématiquement, on fermera un PCA
par un coïl si le diamètre est < à 3mm (Image 15) et par une prothèse si il
est > à 3mm (Image 16). Le choix final est de toute façon guidé par l’aspect
anatomique du canal à l’angiographie initiale.
Il existe une particularité à l’examen clinique des patients ayant un syndrome
d’Eisenmenger secondaire à un large canal artériel. Le shunt artériel est à
l’origine d’une désaturation plus importante aux membres inférieurs par rap-
port aux membres supérieurs. Initialement, cela se traduit cliniquement par
un hippocratisme digital intéressant seulement les orteils (Image 17).
Remerciements à mes collègues les Docteurs
Jean-Christophe Eicher et Abdelkader Touiza.
SESSION d’OCTOBRE 2011
1
/
3
100%