COURS 8 PHYSIOPATHOLOGIE DE L`APPAREIL RESPIRATOIRE

COURS 8
PHYSIOPATHOLOGIE DE L’APPAREIL RESPIRATOIRE (III)
PLAN DU COURS:
I. LE SYNDROME D’APNEE OBSTRUCTIVE DE SOMMEIL
II. LE SYNDROME D’INSUFFISANCE RESPIRATOIRE
III. LE SYNDROME DE DETRESSE RESPIRATOIRE AIGUE
I. LE SYNDROME D’APNEE OBSTRUCTIVE DE SOMMEIL (AOS)
A. DÉFINITION
Le syndrome d’apnée au cours du sommeil est défini par des épisodes récurrents d’interruption (apnée) ou
de réduction (hypopnée) du flux d’air au niveau de l’oropharynx, accompagnés ou non par des micro-
éveils corticaux, avec la fragmentation accentuée du sommeil et la somnolence diurne excessive.
L’apnée
représente l’interruption totale du flux d’air au niveau de l’oropharynx, pour des périodes
supérieures à 10 secondes (pour la majorité des patients les périodes d’apnée ont une durée de 20-30
secondes et elles peuvent arriver à 2-3 minutes)
L’hypopnée représente la réduction du flux d’air au niveau de l’oropharynx de 30-50% pour des
périodes supérieures à 10 secondes accompagné par la réduction de la saturation en oxigène de
l’hémoglobine (SaO2) dans le sang périphérique de plus de 3-4% ou d’un micro-éveil
L’indice apnée/hypopnée (IAH)
représente le rapport entre le nombre des apnées / des hypopnées et
le temps total de sommeil, lui étant suggestif pour l’apnée de sommeil s’il est supérieur à 5/h (5-14 –
forme légère, 15-29 – forme modérée et > 30 – forme sévère)
Le micro-éveil cortical - la réaction d’éveil identifiée sur l’EEG, sans être conscientisée par le patient et
qui apparaît comme une conséquence de l’hypoxémie
B. PATHOGENÈSE:
SASO est caractérisée par la suivante séquence d’évènements qui se répètent 400-500 fois par nuit:
Le collapsus des voies respiratoires supérieures, avec l’installation de l’apnée
L’apnée produit l’hypoxémie 60 mmHg jusqu’au micro-éveil cortical/réveil brusque du sommeil
Après le rétablissement de la perméabilité des voies respiratoires, les patients rendorment (Fig. 1)
DEPARTEMENT III – SCIENCES FONCTIONNELLES
Discipline de PHYSIOPATHOLOGIE
Rue Tudor Vladimirescu, no. 14
300173 Timioara,
Tel/Fax: +40 256 493085

Figure 1. Séquence primaire d’évènements dans l’apnée obstructive de sommeil (AOS).
(Modifié selon Harrison – Principes de Médecine interne, Ed.14, 2003)
C. LES FACTEURS DE RISQUE:
1) L’influence de L’AGE et DU SEXE
entre 30 et 60 ans la prévalence SAOS est supérieure chez les hommes que chez les femmes
chez les hommes il y a une résistance pharyngienne supérieure et une activité déficitaire de la
musculature dilatatrice pharyngienne
chez les femmes, sous l’influence des hormones œstrogènes, les voies aériennes sont plus stables et
qui peuvent être moins collapsées, malgré leur diamètre moindre
l’âge de 60 ans la prévalence SAOS est égale chez les hommes et les femmes et elle augmente
en même temps que l’âge par l’apparition de l’hypotonie de la musculature de l’oropharynx et
la diminution de la sensibilité des chémorécepteurs périphériques à l’hypoxémie
2) L’obésité CENTRALE (tronculaire, androïde)
Est présente au 80% des patients (isolée ou dans le cadre du syndrôme métabolique)
Réduit le calibre des voies respiratoires supérieures par:
L’augmentation de la déposition de graisse dans les tissus mous du pharynx
La compression du pharynx par les masses grasses superficielles du niveau du cou
À retenir!
L’augmentation de la circonférence du cou de plus de 43 cm chez les hommes et de plus de 41 cm chez
les femmes semble se corréler mieux avec l’incidence et la sévérité SAOS que l’obésité en général.
3) Anomalies STRUCTURALES des voies respiratoires supérieures L’hypertrophie adénotonsillaire, la
rétrognatie, la macroglossie Sont plus fréquentes chez les enfants, isolées ou dans le cadre des
syndrômes génétiques (sdr. Down, sdr. Prader-Willi):
4) Anomalies FONCTIONNELLES des voies respiratoires supérieures maladies neuromusculaires qui
affectent le tonus normal de la musculature autant dans l’état de veille que pendant le sommei, ex. la
sclérose latérale amyotrophique, quelques polyneuropathies, mènent à l’hypoventilation nocturne, des
épisodes répétitifs d’apnée/hypopnée et la fragmentation du sommeil
5) La pathologie ENDOCRINE
Pression négative de l’oropharynx
- activité diminuée des muscles des voies
respiratoires supérieures
- cavité pharyngienne petite
-
résistance augmentée au flux
EVENEMENTS PRIMAIRES
Installation du sommeil
APNEE
Hypoxémie / hypercapnie
Eveil
Reprise du flux d’air
Reprise du sommeil

L’acromégalie, l’hypothyroïdie et le sdr. de Cushing déterminent des infiltrations et des œdèmes des
parties molles qui diminuent le calibre des voies respiratoires supérieures
6) La consommation d’ALCOOL réduit le tonus de la musculature pharyngienne et déprime la réponse
d’éveil à la fin de chaque poussée d’apnée (elle augmente la prévalence / la sévérité SAOS)
7) Les facteurs favorisants :
- Le ronflement vibration à haute fréquence des tissus mous du niveau du palais et du pharynx qui
peut aggraver l’étrécissement des voies respiratoires par l’œdème des tissus
- La position couché pendant le sommeil – réduit le diamètre des voies respiratoires supérieures
(l’effet de la gravitation sur la luette, palais et la langue) et augmente la résistance au flux d'air)
D. Conséquences CLINICO-FONCTIONNELLES:
a) CARDIORESPIRATOIRES
1. La réponse pathologique de la stimulation des chémorécepteurs périphériques dans les conditions
d’une hypoxémie 60 mmHg qui ne peut pas être corrigée par l’augmentation de la ventilation
détermine:
bradycardie dans la période d’apnée jusqu’à 30-50 b/min, suivie par la tachycardie jusqu’à 90-120
b/min dans la phase de reprise de la ventilation, responsables de l’apparition de certains troubles de
rythme ventriculaire (par ex. extrasystoles, tachycardie ventriculaire non soutenue) et par le risque
augmenté de mort subite (IAH 20 par heure)
vasoconstriction systémique responsable de l’hypertension artérielle, ischémie du myocarde et
infarctus du myocarde, accident ischémique cérébral
vasoconstriction pulmonaire responsable de l’hypertension pulmonaire chronique
2. L’augmentation de la post-charge du ventricule gauche (déterminée par l’hypertension systémique)
à l’occasion de chaque événement obstructif détermine l’installation / l’aggravation de l’insuffisance
cardiaque gauche chez les patients à maladie cardiaque préexistante
b) NEUROLOGIQUES et COMPORTEMENTALES
1) La perte / fragmentation du sommeil profond et du sommeil paradoxal (avec des rêves, REM), le
sommeil agité et l’hypoxie cérébrale déterminent:
La somnolence diurne excessive dans des situations passives la lecture, regarder la télé ou
dans toutes les activités quotidiennes au fur et à mesure que la maladie évolue
Les troubles d’attention, de mémoire la diminution de la performance professionnelle
Des changements de personnalité irritabilité, poussées d’anxiété, dépression
Céphalée matinale pulsatile
Nycturie
Impotence chez les hommes
Chez les enfants : des faibles performances scolaires, troubles du comportement (agression), les
parasomnies (parler pendant le sommeil, somnambulisme, terreur nocturne)
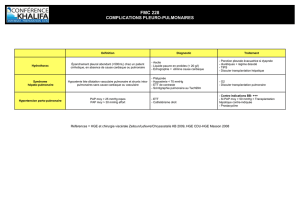
II. LE SYNDROME D’INSUFFISANCE RESPIRATOIRE
A. Définition: l’altération des échanges gazeux pulmonaires caractérisée par la réduction de l’oxygénation du
sang veineux et par l’élimination du dioxyde de carbone
B. Classification:
Selon l’évolution
i. aiguë (heures jours, augmentation aiguë du PaCO2, pH diminué)
ii. chronique (pH plus près du normal ou légérement baissé)
iii. aiguë sur fond chronique (la diminution du PaO2 de 15 mmHg au moins par rapport à la valeur
antérieure)

Selon LES MANIFESTATIONS
i. latente (l’hypoxémie apparaît seulement à l’effort)
ii. manifeste (hypoxémie présente également pendant le repos)
Selon le MECANISME PATHOGENIQUE ET LES VALEURS DES GAZ RESPIRATOIRES au repos:
a) IR de type I ou partielle seulement l’échange pour l’O2 est affecté
i. PaO2 < 60 mmHg (HYPOXÉMIE)
ii. PaCO2 normale ou baissée < 40 mmHg (NORMO- ou HYPOCAPNIE)
b) IR de type II ou globale l’échange pour l’O2 et le CO2 est affecté
i. PaO2 < 60 mmHg (HYPOXÉMIE)
ii. PaCO2 > 50 mmHg (HYPERCAPNIE)
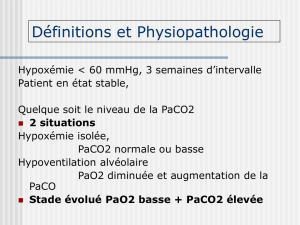
L’ HYPOXEMIE
I. DÉFINITION: la diminution du PaO2 < 60 mmHg
II. MECANISMES PATHOGENIQUES DE L’HYPOXEMIE :
L’insuffisance respiratoire hypoxémique (type I, partielle) est due à:
a. L’altération du rapport ventilation/perfusion VA/Q
b. L’altération de la diffusion des gaz par la membrane alvéolo-capillaire
c. La présence des shunts artério-veineux
L’insuffisance respiratoire hypercapnique / hypoxémique (type II, totale) est due à:
d. La diminution du PO2 dans l’air inspiré
e. L’hypoventilation alvéolaire globale
a. L’altération du RAPPORT VA/Q
Cause:
maladies pulmonaires obstructives pareilles à l’asthme bronchique ou au BPOC
œdème pulmonaire cardiogène et non-cardiogène
Mécanisme PATHOGÉNIQUE: la coexistence des territoires hypoventilés avec des territoires normo- et
hyperventilés
1. Dans les territoires HYPOVENTILÉS (déficit de ventilation par rapport à la perfusion) le rapport VA/Q
est inférieur à 0,8:
Les pressions des gaz respiratoires du sang artériel tendent vers les valeurs du sang veineux,
(PaO2 = 40 mmHg, PaCO2 = 47 mmHg)
Le sang quittant les territoires hypoventilés a une saturation diminuée en O2 (SaO2 97%)
2. Dans les territoires HYPERVENTILÉS (excès de ventilation par rapport à la perfusion) le rapport VA/Q
est supérieur à 0,8:
Les pressions des gaz respiratoires du sang artériel tendent vers les valeurs de l’air atmosphérique
(PaO2 = 130 mmHg, PaCO2 = 0,23 mmHg)
Le sang quittant les territoires hyperventilés a une saturation normale en O2 (SaO2 = 97%)
3. Le sang quittant le poumon a PaO2 < 60 mmHg, PaCO2 normale ou PaCO2 < 40 mmHg et SaO2
97%
La désaturation du sang des territoires hypoventilés ne peut pas être compensée par une
«sursaturation» du sang des territoires hyperventilés parce que la forme en «S» de la courbe de
dissociation de l’oxyhémoglobine (Fig.10.3.1) ne permet pas l’augmentation du SaO2 100%, quelle
que soit l’augmentation du PaO2
L’accumulation du CO2 des territoires hypoventilés est compensée par l’élimination augmentée de
CO2 dans les zones hyperventilées parce que la relation entre la concentration de CO2 et PCO2 du
plasma est linéaire
Observation!

L’administration d’O2 corrige rapidement l’hypoxémie par la normalisation du SaO2% des zones
hypoventilées (l’augmentation du gradient de pression alvéolo-capillaire de l’O2 force la diffusion de l’O2
par la membrane alvéolo-capillaire)
b. L’altération de la DIFFUSION DES GAZ PAR LA MEMBRANE ALVÉOLO-CAPILLAIRE
Causes:
a) L’augmentation de la longueur de la voie de diffusion et / ou la réduction de la perméabilité de la
membrane alvéolo-capillaire
œdème pulmonaire
maladies pulmonaires interstitielles (fibrose pulmonaire)
inflammations pulmonaires (ARDS, pneumonies / bronchopneumonies)
b) La diminution de la surface de diffusion alvéolo-capillaire
- Des résections pulmonaires
- Emphysème pulmonaire
Mécanisme PATHOGÉNIQUE: les processus pathologiques qui affectent la diffusion alvéolo-capillaire ont
des conséquences sur le transfert O2, lorsque le transfert de CO2 reste le même (le coefficient de diffusion
du CO2 est 20 fois supérieur à celui d’O2)
Observation!
La diminution PaO2 des troubles de diffusion alvéolo-capillaire est aggravée par la respiration dans
l’atmosphère pauvre en O2 et l’effort physique, mais elle est corrigée par l’administration d’O2
(l’augmentation du gradient de pression alvéolo-capillaire de l’O2 force la diffusion de l’O2 par la membrane
alvéolo-capillaire)
c. La présence des SHUNTS ARTERIO-VEINEUX
Causes:
a) Shunts PULMONAIRES
Par des voies vasculaires anatomiques anormales embolie pulmonaire sévère, atélectasie,
pneumothorax
Par des voies vasculaires pathologiques anévrismes artério-veineux
b) Shunts EXTRAPULMONAIRES cardiopathies congénitales à shunt droite-gauche (ex, tétralogie
Fallot)
Mécanisme PATHOGÉNIQUE:
Le passage d’une fraction du sang veineux (PO2 = 40 mmHg) directement dans la circulation artérielle
systémique (PO2 =100 mmHg) détermine la diminution du PaO2 60 mmHg et du SaO2 97%
L’hypoxémie induit l’hyperventilation reflexe qui compense l’excès de CO2 apporté par le sang
veineux shunté, si bien que le PaCO2 est 40 mmHg
Observation!
L’administration d’O2 ne corrige pas l’hypoxémie parce que la désaturation donnée par le mélange du sang
veineux avec le sang artériel ne peut pas être compensée par une «sursaturation» du sang artériel (la
forme en "S" de la courbe de dissociation de l’HbO2 ne permet pas l’augmentation du SaO2 100% même
si la pression O2
augmente). Autrement dit, l’augmentation du gradient de pression alvéolo-capillaire de
l’O2 ne peut pas forcer la diffusion O2 par la membrane alvéolo-capillaire.
d. La diminution du PO2 dans L’AIR INSPIRÉ
Causes:
La respiration à grande altitude (la diminution de la pression atmosphérique)
L’inhalation de gaz toxiques ou d’air vicié à contenu réduit d’O2
e. Hypoventilation ALVEOLAIRE GLOBALE
Causes:
 6
7
8
9
10
11
6
7
8
9
10
11
1
/
11
100%