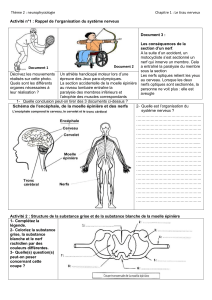
Syndrome de Steele, Richardson et Olszewski - E

Syndrome de Steele, Richardson et
Olszewski
La maladie de Steele, Richardson et Olszewski est une pathologie dégénérative du cerveau
décrite en 1963 qui survient dans la même tranche d’âge que la maladie de Parkinson, et touche
l'homme ou la femme entre 50 et 70 ans. D’autre part, c’est dans ce groupe de parkinsoniens que
de nombreux cas de patients atteints par ce syndrome ont été rencontrés.
Des virus, qui ne sont pas tous identifiés, entraînent des maladies ayant en commun une phase de
longue période muette qui dure de quelques mois à quelques années, période qui s'étale du
moment où le virus pénètre dans l'organisme du patient jusqu’à celui où apparaissent les premiers
symptômes de la maladie.
Il existe d'autres caractéristiques des virus lents, comme l'atteinte d'un seul organe (comme le
système nerveux central), l'atteinte d'une seule espèce animale, et une évolution progressive
vers la mort.
La cause exacte de cette maladie est inconnue, et même si certains virus lents ont été suspectés,
les essais de transmission faits aux singes par inoculation intracérébral de broyats de cerveaux
n’ont rien donné (on injecte à des singes des mélanges finement broyés de cerveau de singes eux-
mêmes atteints par des virus lents).
Le début se fait en général après 60 ans. On constate tout d'abord des anomalies concernant les
yeux avec en particulier : une atteinte marquée de la verticalité des mouvements oculaires, une
paralysie du regard vers le bas et une hypertonie c'est-à-dire une exagération de la tonicité
musculaire de la nuque qui est en extension. Quelquefois le patient a des difficultés à monter ou
à descendre les escaliers.
Les autres symptômes sont
Regard figé.
Difficulté à regarder vers le bas.
Troubles de l’équilibre.
Troubles de la marche.
Chutes inexpliquées.
Impossibilité d'effectuer des mouvements avec l'œil, et plus spécifiquement les mouvements
permettant de regarder de haut en bas.Hypertonie musculaire (tonicité trop importante des
muscles) du tronc, à l'origine d'une hyperextension de la colonne vertébrale (dos courbé en
arrière) et de la nuque.
Acinésie faciale (absence de mouvements du visage).
L'évolution est différente selon les patients. Elle est émaillée de complications (pneumonie,
traumatismes, fracture etc.).

De façon générale celle-ci est péjorative étant donnée que la survie moyenne ne dépasse pas
quelques années.Au cours de la paralysie supranucléaire progressive certaines complications sont
susceptibles de survenir :
Atteinte pulmonaire à type de pneumonie par aspiration.
Traumatisme crânien.
Fractures dues aux chutes.
Les signes évoluent pendant des mois voire des années jusqu’à ce que le patient devienne
quasiment anarthrique (ne puisse plus bouger), avec une perte complète du contrôle du
mouvement des yeux de manière volontaire et une impossibilité à mobiliser de plus en plus
grande son tronc (rigidité axiale sévère).
L’évolution se fait en quelques années vers un syndrome pseudo-bulbaire. Le syndrome pseudo
bulbaire est secondaire à des lésions bilatérales (des deux côtés) de plusieurs zones de
l’encéphale (système nerveux contenu dans le crâne). La capsule interne (amas de substance
grise contenu dans la substance blanche du cerveau) . Le cortex (couche de cellules grises
recouvrant la substance blanche à la périphérie du cerveau).
Le syndrome de paralysie pseudo bulbaire se caractérise par des troubles qui simulent (qui
ressemblent à) une atteinte du bulbe lui-même (le bulbe étant la région de l’encéphale située en
avant du cervelet, juste au-dessus de la moelle épinière). Ces symptômes sont une paralysie des
muscles de la déglutition (le patient ne peut plus avaler), de la phonation (le patient ne peut plus
émettre des sons), de la langue et des lèvres.
L'évolution s'accompagne également d'un affaiblissement de l'intellect (troubles cognitifs).
Le nombre de neurones diminue très rapidement dans certaines zones du cerveau (noyau des
nerfs crâniens) qui sont des îlots de substance grise dans la substance blanche. La démence est
variable suivant les individus.
Le traitement est à peu près inefficace.
Des médicaments visant à lutter contre le syndrome de Parkinson ont été essayés, mais
n’entraînent pas de grand bénéfice.
Certains médecins utilisent la lévodopa et une association lévodopa et anticholinergiques qui sont
des médicaments utilisés chez les parkinsoniens, susceptibles de diminuer certains symptômes
cités ci-dessus.
L’idasoxan est susceptible de favoriser la transmission des messages nerveux entre les neurones
(cellules nerveuses) et plus particulièrement les neurones fonctionnant avec le
neurotransmetteur appelé noradrénaline. Ces neurones sont plus spécifiques d’un mécanisme
permettant une amélioration des mouvements qui est obtenue chez certains patients.
D'autre médicament qui est également été essayé et tel que le zolpidem ou l'amantadine
1
/
2
100%