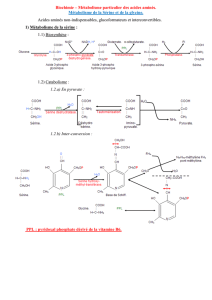
Métabolisme de l homocystéine

57
Le Courrier de l’Arcol (1), n° 2, juin 1999
Mise
au point
homocystéine est un acide aminé
soufré, homologue supérieur de la
cystéine. Il présente la particularité de ne
pas être incorporé dans les protéines, et il
existe dans l’organisme sous différentes
formes en équilibre d’oxydoréduction :
homocystéine, homocystine (constituée de
deux résidus d’homocystéine réunis par
un pont disulfure), disulfure mixte homo-
cystéine-cystéine (1-3).
Dans le plasma, 70 % de l’homocystéine
circule sous une forme liée aux protéines
par des ponts disulfure. L’homocystéine dite
“libre” est essentiellement représentée par
les disulfures mixtes homocystéine-cystéine
(figure 1, page 58).
Métabolisme
de l’homocystéine
(4)
L’homocystéine est formée au cours du
métabolisme de la méthionine, dont l’ori-
gine est principalement alimentaire (figure 2,
page 59). La méthionine est utilisée par
l’organisme sous sa forme activée, la S-adé-
nosyl-méthionine, au cours des réactions de
méthylation catalysées par les méthyl-trans-
férases. La fixation des groupements méthyl
sur leur substrat (acides nucléiques, pro-
téines, catécholamines par exemple) forme
la S-adénosyl-homocystéine, à partir de
laquelle l’homocystéine est libérée par
hydrolyse.
L’homocystéine peut être métabolisée de
deux façons distinctes :
•
La première possibilité est la voie de la
transsulfuration, qui conduit à la formation
de cystéine. Ce métabolisme, surtout actif
dans le foie, le rein, le pancréas et le
cerveau, est irréversible et permet de cata-
boliser l’homocystéine présente en excès.
Deux réactions constituent cette voie
métabolique. La première, qui forme la
cystathionine par réaction avec la sérine, est
catalysée par la cystathionine synthase. La
seconde, catalysée par la cystathionase,
libère l’homosérine et la cystéine, dont le
catabolisme final est la formation de sul-
fates. L’activité de ces deux enzymes est
dépendante de la vitamine B6 (phosphate de
pyridoxal).
•
La seconde possibilité est la reméthylation
en méthionine, qui constitue un système de
conservation de celle-ci et qui peut être
assurée par deux voies métaboliques
distinctes :
– une voie mineure, localisée essentielle-
ment dans le foie, catalysée par la bétaïne-
homocystéine méthyltransférase : celle-ci
utilise la bétaïne comme donneur de méthyl
et libère la diméthyl-glycine ;
– une voie majeure, ubiquitaire, catalysée
par la méthionine synthase, dont le cofac-
teur est la vitamine B12.
Cette enzyme synthétise la méthionine en
fixant sur l’homocystéine un groupement
méthyl fourni par le 5-méthyl-tétrahydrofo-
late (5-méthyl-THF). Le tétrahydrofolate
(THF) libéré au cours de cette réaction est
au centre d’un cycle de réactions passant
par le 5-10-méthylène-THF, puis le
➬L’homocystéine est un acide aminé dérivé du métabolisme
de la méthionine, qui n’est pas incorporé dans les protéines.
➬Elle peut être métabolisée de deux façons : soit par transsulfuration,
conduisant à la formation de cystéine, soit par reméthylation en
méthionine.
➬Le métabolisme de l’homocystéine est dépendant de l’apport
en plusieurs facteurs vitaminiques : vitamines B6 et B12, acide folique.
➬Les hyperhomocystéinémies peuvent être provoquées par
des anomalies soit de la transsulfuration, soit de la reméthylation.
➬Leur cause peut être soit génétique (déficits enzymatiques),
soit environnementale (carences vitaminiques).
➬L’exploration du métabolisme de l’homocystéine comporte,
outre le dosage de l’homocystéine circulante totale, des dosages
de vitamines, un test de charge en méthionine, le dépistage
de mutations des gènes codant les enzymes impliquées.
➬La mutation associée aux hyperhomocystéinémies modérées,
la plus recherchée en pratique courante, est celle du gène
de la MTHFR en position 677.
➬La double origine des anomalies (génétique et acquise) permet
d’envisager des interventions thérapeutiques sur le versant
environnemental (supplémentations vitaminiques).
* Laboratoire central de biochimie,
hôpital Robert-Debré, Reims.
Métabolisme de l’homocystéine
Ph. Gillery*
P
O
I
N
T
S
F
O
R
T
S
L
L’
’

58
Le Courrier de l’Arcol (1), n° 2, juin 1999
5-méthyl-THF. Il est apporté à l’organisme
par l’alimentation, sous la forme d’acide
folique rapidement réduit en THF. Le
5-méthyl-THF est recyclé en permanence
sous l’action de l’enzyme méthylène-
tétrahydrofolate-réductase (MTHFR), qui
exerce donc une action indirecte mais déter-
minante dans la reméthylation de l’homo-
cystéine.
Mécanismes
des hyperhomocystéinémies
L’exposé des voies de métabolisme de l’ho-
mocystéine et de la méthionine permet de
déduire que les hyperhomocys-
téinémies sont liées à des
troubles affectant soit la trans-
sulfuration, soit la reméthyla-
tion. En effet, les augmenta-
tions d’apport ou de production
de méthionine ne sont pas
capables d’induire par elles-
mêmes une hyperhomocys-
téinémie, hormis lors d’une
charge massive, utilisée au
cours de tests diagnostiques, à
des concentrations très supé-
rieures aux apports alimen-
taires.
Les causes des hyperhomocys-
téinémies sont soit d’origine
génétique, soit d’origine envi-
ronnementale.
Les causes génétiques
•
Le déficit en cystathionine
synthase représente le déficit
enzymatique classique condui-
sant à l’homocystinurie. Sa
prévalence est de 1/200 000, et
sa transmission autosomique
récessive. Plus de trente mu-
tations différentes de la cysta-
thionine synthase ont été identi-
fiées. Les formes homozygotes
conduisent à l’homocystinurie
typique, avec hyperhomocystéi-
némie et hyperméthioninémie.
Les signes cliniques associent
des troubles neurologiques et
oculaires, des anomalies sque-
lettiques et des signes d’athéro-
sclérose précoce. Les formes
hétérozygotes, présentes dans
moins de 1 % de la population générale,
s’accompagnent rarement d’hyperhomocys-
téinémie (2 ,5).
•
Les enzymes du cycle de méthylation peu-
vent également être affectées. Leur atteinte
peut entraîner, plus rarement que dans le cas
précédent, d’authentiques homocystinuries.
Plus souvent, elles sont associées à des
hyperhomocystéinémies modérées.
Les principales anomalies génétiques de
cette voie touchent le gène de la MTHFR.
Dans les formes sévères, rares, l’homocys-
téine est augmentée dans le plasma et l’urine,
la méthionine plasmatique est diminuée, et
divers troubles, notamment neurologiques
et cardiovasculaires, sont notés. L’activité
résiduelle MTHFR est faible, et différentes
mutations responsables de ces tableaux ont
été identifiées (6).
Par ailleurs, un polymorphisme génétique
courant de ce gène a été identifié : la substi-
tution C ➝T du nucléotide 677, qui conduit
à l’incorporation d’une valine à la place
d’une alanine dans la protéine (7). Cette
modification crée un site de clivage Hinf I,
qui permet sa mise en évidence après ampli-
fication de l’ADN par PCR. La mutation
s’accompagne d’une diminution de l’activité
et d’une augmentation de la thermolabilité de
l’enzyme codée. Il s’agit d’une mutation fré-
quente dans les populations occidentales :
environ 12 % d’homozygotes et 40 à 45 %
d’hétérozygotes (8). D’autres mutations du
gène de la MTHFR, associées à des déficits
modérés de l’activité enzymatique et d’éven-
tuel intérêt clinique, ont été décrites (9).
En pratique courante, la mutation C677T est
la mutation le plus souvent recherchée. Son
association avec les pathologies athéro-
thrombotiques est toujours discutée (10).
•
Enfin, des maladies génétiques rares du
métabolisme de la vitamine B12 peuvent
être responsables d’hyperhomocystéinémie.
Elles s’accompagnent d’anémie mégaloblas-
tique, de troubles neurologiques, et parfois
de manifestations thromboemboliques (2).
Les causes environnementales
Trois facteurs vitaminiques sont, comme on
l’a décrit plus haut, impliqués dans le méta-
bolisme de l’homocystéine :
–la vitamine B12, cofacteur de la méthio-
nine synthase,
–l’acide folique, précurseur direct du THF,
– la vitamine B6, cofacteur des réactions de
transsulfuration.
Les deux premiers facteurs interviennent
dans la voie de reméthylation de l’homocys-
téine.
Les carences en ces facteurs, principalement
dues à une insuffisance de leur apport alimen-
taire, sont directement responsables d’hyper-
homocystéinémie, généralement modérée.
Les carences en acide folique, et à un moindre
degré en vitamine B12, sont le plus souvent
associées à l’hyperhomocystéinémie.
D’autres causes environnementales, impli-
quant à divers degrés ces métabolismes ou
influant sur le métabolisme rénal de
l’homocystéine, sont à l’origine d’hyper-
homocystéinémies (2) :insuffisance rénale,
cancer, maladies diverses comme le psoria-
sis, traitements médicamenteux (métho-
trexate, anticonvulsivants, NO).
Mise
au point
H3C–S–CH
2–CH
2–CH–COOH Méthionine
NH2
HS – CH2–CH
2–CH–COOH Homocystéine
NH2
HS – CH2–CH–COOH Cystéine
NH2
CH2–CH
2–CH–COOH
Homocystine
S NH2
S
CH2–CH
2–CH–COOH
NH2
CH2–CH
2–CH–COOH
Disulfure mixte
S NH2homocystéine-cystéine
S
CH2–CH–COOH
NH2
CH2–CH
2–CH–COOH
Homocystéine
S NH2liée à une protéine
S
Protéine
Figure 1. Structure des acides aminés impliqués dans le
métabolisme de l’homocystéine et des différentes formes
de l’homocystéine dans l’organisme.

59
Le Courrier de l’Arcol (1), n° 2, juin 1999
Bases métaboliques
des explorations biologiques
En clinique, l’étude du métabolisme de
l’homocystéine peut, outre le dosage de
l’homocystéine totale, être complétée par
des examens destinés à l’exploration des
différentes voies décrites plus haut :
–le test de charge en méthionine, qui consiste
àadministrer par voie orale 0,1 g de méthio-
nine par kg de masse corporelle, générale-
ment dans un jus d’orange (les dosages
d’homocystéine plasmatique sont le plus
souvent effectués à 0, 2 , 4 et 6 heures) ;
–les dosages des facteurs vitaminiques
impliqués ;
–la détection de polymorphismes du gène
de la MTHFR (C677T en premier lieu).
Le test de charge, initialement utilisé pour
détecter les sujets hétérozygotes pour le
déficit en cystathionine synthase, permet de
mettre en évidence des anomalies, même
modérées, de la voie de transsulfuration (3).
Des travaux récents suggèrent cependant
que ce test puisse également révéler des
anomalies de la voie de reméthylation.
Nous ne discuterons pas ici des résultats et
des valeurs seuils retenus en clinique, car la
plupart de ces tests ne sont pas encore stan-
dardisés. Ce sujet fait l’objet d’un autre
article de cette revue.
Conclusion
Le métabolisme de l’homocystéine peut être
affecté par des anomalies tant génétiques
qu’environnementales. Différentes investiga-
tions biologiques en permettent un diagnostic
de plus en plus fin. Les diverses carences
vitaminiques génératrices d’hyperhomocys-
téinémie suggèrent la possibilité d’une prise
en charge thérapeutique effective, et des
études prospectives d’intervention sont
menées à l’heure actuelle par plusieurs
équipes. ■
Références
1. Jacobsen D.W. Homocysteine and vitamins in
cardiovascular disease. Clin Chem 1998 ; 44 :
1833-43.
2. Bakker R.C., Brandjes D.P.M. Hyperhomocysteinemia
and associated disease. Pharm World Sci 1997 ; 19 :
126-32.
3. Refsum H., Ueland P.M., Nygard O., Vollset S.E.
Homocysteine and cardiovascular disease. Annu
Rev Med 1998 ; 49 : 31-62.
4. Finkelstein J.D. The metabolism of
homocysteine : pathways and regulation. Eur J
Pediatr 1998 ; 157 : S40-4.
5. Mudd S.H., Levy H.L., Skovby F. Disorders of
transsulfuration. In : Scriver C.R., Beaudet A.L.,
Sly W.S. et coll. (eds) : The metabolic and molecular
basis of inherited diseases. New York, Mc Graw-
Hill, 1995 : 1279-327.
6. Kluijtmans L.A.J., Wendel U., Stevens E.M.B.,
Van den Heuvel L.P.W.J., Trijbels F.J.M., Blom H.J.
Identification of four novel mutations in severe
methylenetetrahydrofolate reductase deficiency. Eur
J Hum Genet 1998 ; 6 : 257-65.
7. Frosst P., Blom H.J., Milos R., Goyette P.,
Sheppart C.A., Matthews R.G., Boers G.J.H., Den
Heijer M., Kluijtmans L.A.J., Van den Heuvel L.P.,
Rozen R. A candidate genetic risk factor for vascular
disease : a common mutation in methylenetetrahy-
drofolate reductase. Nature Genet 1995 ; 10 : 111-3.
8. Rozen R. Genetic predisposition to hyperhomocy-
steinemia : deficiency of methylenetetrahydrofolate
reductase (MTHFR) Thromb Haemost 1997 ; 78 :
523-6.
9. Weisberg I., Tran P., Christensen B., Sibani S.,
Rozen R. A second genetic polymorphism in methy-
lenetetrahydrofolate reductase (MTHFR) associated
with decreased enzyme activity. Mol Genet Metab
1998 ; 64 : 169-72.
10. Fletcher O., Kessling A.M. MTHFR association
with arteriosclerotic vascular disease ? Hum Genet
1998 ; 103 : 11-21
.
Mise
au point
Sérine
Glycine
N5-N10 méthylène-
THF
Diméthyl-
glycine
S-ad-Met
S-ad-Hcy
Substrat
Adénosine
Substrat
méthylé
ATP
bétaïne
MTHFR
Méthionine
synthase
Sérine hydroxyméthyltransférase
BHMT
5-méthyl-THF
Cystathionine
Cystathionine synthase
Méthionine-adénosyl-transférase
Méthyltransférase
Adénosyl homocystéinase
Cystathionase
Cystéine
Réactions de
reméthylation
de l'homocystéine
Réactions
de méthylation
Réactions
de transsulfuration
Acide folique
Vit B12
Méthionine
Vit B6
Vit B6
THF
Homocystéine
Protéines
Figure 2. Voies métaboliques impliquant l’homocystéine.
BHMT : bétaïne-homocystéine méthyltransférase. S-ad-Met : S-adénosyl-méthionine. S-ad-Hcy : S-adénosyl-homocystéine.
THF : tétrahydrofolate. MTHFR : méthylène-tétrahydrofolate-réductase.
Les constituants apportés par l’alimentation sont indiqués sur fond bleuté.
1
/
3
100%