Anomalies chromosomiques

Cours 5 : Méiose – Fécondation – Cycle biologique –
Anomalies chromosomiques
Partie 3 : Les anomalies chromosomiques
Rappel : organisation des chromosomes :
- Le centromère, parfois appelé constriction primaire,
divise le chromosome en 2 régions, le bras court p et le
bras long q.
- Les télomères sont les extrémités terminales des
chromosomes au niveau des bras courts et des bras longs.
- Les constrictions secondaires sont présentes sur les
chromosomes acrocentriques (type de chromosomes dont
le centromère se situe à l'extrémité du bras q)
- Les satellites qui se présentent sous forme de sphères de
taille variable sont situés à l’extrémité du bras court des
chromosomes acrocentriques.
- Les chromosomes médiocentriques ont un
centromère situé au milieu du chromosome.
- Les chromosomes métacentriques et
submétacentriques ont un centromère légèrement décalé
vers l’une des extrémités du chromosome, ce qui donne 2
bras de longueur presque égale.
- Les chromosomes télocentriques ou
subtélocentriques ont un bras court très petit.
- Les chromosomes acrocentriques ont un centromère
qui semble se situer pratiquement à une extrémité et
possèdent des satellites. (ex : chromosomes 13, 14, 15, 21,
22).

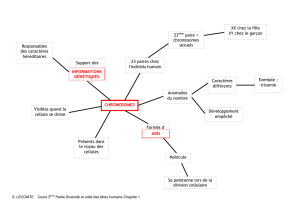
Les anomalies chromosomiques regroupent toutes les anomalies de nombre ou de structure d’un (ou plusieurs)
chromosome(s) dans un génome. Les anomalies (ou aberrations) chromosomiques peuvent s'observer de manière
constitutionnelle (elles sont alors présentes dès la naissance) soit de manière acquise au cours de processus malins
(ils ne sont observés alors qu'au niveau des cellules tumorales). Elles résultent d'un accident survenant soit au cours
de la méiose, soit au cours d'une mitose. Elles peuvent impliquer un ou plusieurs chromosomes.
On reconnaît par ailleurs les anomalies dites homogènes (quand toutes les cellules examinées portent
l'anomalie, en lien avec une anomalie de ségrégation des chromosomes à la méiose) et les anomalies en mosaïque
quand une fraction seulement des cellules est anormale (sont le résultat d'un évènement post-zygotique donc après
la fécondation, il s’agit d’erreur de ségrégation des chromosomes lors d'une mitose chez un embryon à caryotype
normal ou de la correction spontanée d'une anomalie).
Leurs conséquences sont variables en fonction du remaniement considéré. En règle générale, les remaniements
dits équilibrés (c'est-à-dire sans perte ni gain de matériel génétique) n'ont habituellement pas de conséquence pour
le sujet porteur alors que les remaniements déséquilibrés se traduisent par des manifestations cliniques d'autant
plus graves que la perte ou le gain de matériel est plus important.
I- Anomalie de nombre = nombre de chromosomes anormal
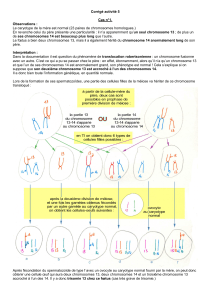
La non-disjonction conduit à l’aneuploïdie
La non-disjonction des chromosomes correspond à l'absence de séparation des chromosomes homologues en
anaphase de méiose 1 ou à la non-disjonction des chromatides sœurs en méiose 2.
Principe de la non-disjonction en 1ère et 2ème division de méiose

Cette anomalie de fonctionnement crée soit des gamètes avec un chromosome surnuméraire (trisomie), soit
des chromosomes absents (monosomie). Ce sont des cellules avec une anomalie du nombre de chromosomes
appelées cellules aneuploïdes. On dit que la non-disjonction conduit à l’aneuploïdie.
La plupart des anomalies ne permettent pas le développement de l'embryon et donne lieu à des avortements
spontanés.
Ces anomalies sont fréquentes (6 à 25%), elles concernent toutes les paires de chromosomes et les monosomies
sont statistiquement aussi fréquentes que les trisomies.
Quelques anomalies permettent de mener à terme une grossesse :

Une trisomie correspond à la présence d’un chromosome supplémentaire. Le nombre de chromosomes est donc
de 47 et non plus de 46. Tous les chromosomes peuvent être impliqués, mais seulement trois trisomies
autosomiques sont viables à l’état homogène dans l’espèce humaine :
● Trisomie 21 (Syndrome de Down) : 47, XX ou 47, XY
● Trisomie 13 (Syndrome de Patau) : 47, XX ou 47, XY
● Trisomie 18 (Syndrome d'Edwards) : 47, XX ou 47, XY
D’autres trisomies peuvent être observées en mosaïque, pour le 8 ou le 9 par exemple.
Le gain d’un chromosome peut également concerner les gonosomes :
● Trisomie X (Syndrome Triple X) : 47, XXX
● Le syndrome de Klinefelter : L'individu possède deux chromosomes X et un chromosome Y (XXY) : 47, XXY
● Le Syndrome de Jacob : l'individu possède un chromosome Y en double exemplaire, et un chromosome X (XYY) :
47, XYY
Le syndrome de Klinefelter
• Cytogénétique :
- Dans 85 % des cas, l'anomalie est homogène : 47, XXY dans toutes les cellules.
- Autres cas sont des mosaïques 47, XXY/46,XY ou 47XXY/46XX
• Fréquence : 1/1.000 garçons (mosaïques comprises) - Pas de rôle de l'âge parental
• Clinique :
Signes principaux :
- Atrophie testiculaire
- Gynécomastie
- Stérilité
- Pas de dysmorphie importante
• Diagnostic posé :
- à la puberté le plus souvent
- parfois pour stérilité
- rarement à la naissance, rarement chez le jeune enfant (parfois pour malformation non spécifique des
organes génitaux externes).
• Phénotype :
- Atrophie testiculaire : constante - testicules petits, mous contraste ; avec scrotum de taille et de
pigmentation normale.
- Verge le plus souvent normale, parfois hypoplasique
- Gynécomastie : n'est pas constante (1/3 à 1/4 des cas) ; apparaît vers 12-13 ans, volume modéré, au début
asymétrique.
- Autres anomalies des caractères sexuels secondaires : pilosité rare, répartition gynoïde des graisses
- Stérilité : azoospermie pratiquement constante.
- Morphologie du sujet : variable. Soit sujets longilignes, avec membres longs, soit sujets de morphologie
masculine normale (sujets parfois même de petite taille)
- Développement intellectuel normal dans la majorité des cas ; cependant certains sujets peuvent présenter
des difficultés scolaires et d'inadaptation sociale ou des troubles psychotiques.

Caryotype 47, XXY
Le syndrome de Down (trisomie 21)
La trisomie 21 est la plus fréquente des anomalies de répartition des chromosomes. Elle est associée à une série
de signes cliniques constituant le syndrome de Down. Le cas le plus courant est une mauvaise répartition du
chromosome 21 lors de la méiose 1 entraînant un gamète possédant 2 chromosomes 21 au lieu de deux. Par la suite,
si ce gamète effectue la fécondation, l'individu en résultant possèdera donc 3 chromosomes 21 au lieu de deux dans
son caryotype. On parle de trisomie libre.
D’autres cas de trisomie 21 consistent en une translocation d'un fragment de chromosome 21 sur le
chromosome 14. On parle alors de trisomie 21 par translocation (par opposition à la trisomie 21 libre où le
chromosome surnuméraire est libre).
Il se peut également qu'il y ait une erreur lors la première mitose de la cellule œuf. Ainsi, un des deux
chromosomes 21 ne se sépare pas lors de l'anaphase et va en entier dans une des deux cellules filles, causant des
divisions entraînant des cellules à 3 chromosomes 21.
Un individu trisomique 21 peut donc présenter trois chromosomes 21 distincts ou seulement deux chromosomes
21 libre et une partie de chromosome 21 fusionnée avec le chromosome 14 ou avec un autre chromosome 21 (ici on
dit translocation robertsonienne).
Cytogénétique :
- 95% des cas d'une trisomie libre (Chromosome 21 surnuméraire avec formule 47,XX,+21 ou 47,XY).
- autres cas : trisomie 21 par translocation, le chromosome 21 surnuméraire étant transloqué sur un autre
chromosome acrocentrique, 14 le plus souvent.
Fréquence : 1 pour 700 naissances vivantes ; le risque augmente en fonction de l'âge maternel.
Diagnostic :
- diagnostic prénatal : étude du caryotype foetal
- chez le nouveau-né sur l'association Hypotonie (faible tonus musculaire) + dysmorphie
 6
7
8
9
10
11
12
13
6
7
8
9
10
11
12
13
1
/
13
100%