Télécharger l`article au format PDF

S 858
L’Encéphale, 2006 ;
32 :
858-60, cahier 3
Le parcours du médicament, de la recherche clinique
à la commercialisation. Contraintes, embûches, délais et coûts
I. GIRI
(1)
, F. ROUILLON
(1)
(1) Hôpital Sainte-Anne, Service du Pr Guelfi, clinique des maladies mentales et de l’Encéphale (CMME), 100, route de la Santé,
75674 Paris 14.
LE PARCOURS DU MÉDICAMENT
DANS L’ENTREPRISE PHARMACEUTIQUE
Le parcours du médicament au sein de l’entreprise est
de plus en plus long : il faut compter désormais 14
ans
pour qu’une molécule synthétisée par une firme obtienne
l’Autorisation de Mise sur le Marché
(figure 1)
. De même,
les coûts de développement d’une molécule ont considé-
rablement augmenté : ils étaient d’environ 100
millions de
dollars il y a 20
ans, ils dépassent aujourd’hui 800
millions
de dollars. Ces coûts conduisent souvent les firmes à con-
clure des partenariats, parfois complexes et intriqués, afin
de limiter les risques en cas d’échec d’une molécule, la
probabilité de conduire une molécule jusqu’à l’AMM étant
particulièrement limitée lors des phases précoces du
développement : ainsi, 70
% des dépenses de recherche
et développement financent des échecs…
Le budget recherche et développement des grandes fir-
mes est consacré pour environ 15
% à la recherche fon-
damentale, pour environ un tiers à la recherche appliquée,
et pour plus de la moitié au développement. Pour un médi-
cament dont le dossier d’AMM est déposé, 5
000
à 10
000
molécules ont été initialement criblées, et 250 sont arri-
vées au stade du développement préclinique
(figure 2)
.
LES EXIGENCES ADMINISTRATIVES
Les essais cliniques sont fortement encadrés par de
nombreuses exigences médico-administratives. Ils doi-
vent être conformes aux bonnes pratiques cliniques, rece-
voir un avis de conformité par rapport à la loi Huriet, et
recevoir, conformément aux directives européennes,
l’autorisation de l’Agence du médicament. Ces exigences
peuvent poser des problèmes de délai, par exemple lors-
que la France prend du retard pour obtenir l’autorisation
de participer à un essai multicentrique européen, qui ris-
que alors d’être mené sans elle.
Le nombre de patients qui doit être inclus dans les
essais cliniques pour soumettre un dossier a également
considérablement augmenté : il est par exemple passé
entre
1980 et
1995 d’environ 1
500 à plus de 4
000, tandis
que durant la même période, le nombre d’essais cliniques
par dossier passait d’une moyenne de 30 à près de 70.
FIG. 1. —
Une aventure longue…
Recherche
identification
de la cible
Durée
(années)
2,5 3 1 6 1,5
14 ans
Recherche
candidat
médicament
Développement
pharmaceutique
& préclinique
Développement
clinique (PI, II, III)
AMM
FDA
approval

L’Encéphale, 2006 ;
32 :
858-60, cahier 3 Le parcours du médicament, de la recherche clinique à la commercialisation
S 859
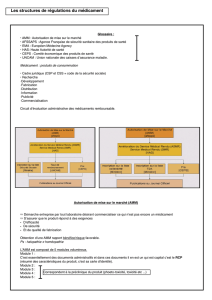
L’Autorisation de Mise sur le Marché
La procédure d’obtention de l’AMM a été codifiée au
niveau européen, avec la mise en place d’une procédure
centralisée, obligatoire depuis novembre
2005 pour les
médicaments concernant le SIDA, le cancer, le diabète et
les maladies neurodégénératives, et facultative pour les
autres innovations. Cette procédure centralisée implique
le recours à des procédures de reconnaissance mutuelle
par chacun des États.
La réponse à une demande d’AMM doit être faite dans
les 210
jours suivant le dépôt de la demande ; l’AMM est
valable durant cinq ans. Les comptes rendus de l’Agence
sont accessibles au public. L’AMM est délivrée en France
par l’Afssaps, après avis de la Commission d’AMM. Lors-
que des exigences de santé publique le justifient, certains
produits peuvent obtenir une Autorisation Temporaire
d’Utilisation (ATU) en attendant l’obtention de l’AMM.
Récemment, de nouvelles évolutions sont apparues
concernant l’AMM. Aux évaluations antérieures s’est ajou-
tée l’évaluation de la Valeur Thérapeutique Ajoutée ; le rap-
port bénéfice/risque doit être réévalué au bout de 5
ans ;
il est nécessaire d’établir un plan de gestion des risques ;
la possibilité est donnée de retirer un médicament du mar-
ché si le rapport bénéfice/risque n’est pas favorable.
L’avis de la Commission de la Transparence
L’AMM permet de commercialiser le produit en officine,
mais n’implique pas la possibilité d’un remboursement.
Elle permet également une prescription initiale hospi-
talière ; pour cela, il faut un agrément aux collectivités.
Pour obtenir le remboursement et un prix, la molécule
doit être évaluée par la Commission de la Transparence,
rattachée à la Haute Autorité de Santé. Celle-ci évalue le
Service Médical Rendu (SMR), qui prend en compte le
rapport bénéfice/risque et la place du produit dans la stra-
tégie thérapeutique, en particulier en fonction de la gravité
de la maladie. Ce SMR est évalué selon 4 grades : insuf-
fisant, faible, modéré, important. Il est également
demandé à la Commission de la Transparence une éva-
luation de l’Amélioration du Service Médical Rendu
(ASMR), évaluation comparative, notée de 1 à 5, qui vient
compléter l’évaluation dans l’absolu du SMR.
SMR et ASMR sont délivrés par indication, et peuvent
concerner certaines populations-cible.
Après la Commission de la Transparence
L’avis de la Commission de la Transparence est trans-
mis au Ministre de la Santé qui décide du remboursement,
à l’UNCAM (Union Nationale des Caisses d’Assurance-
Maladie) qui décide du taux de remboursement, au CEPS
(Comité Économique des Produits de Santé) qui fixe le
prix.
Le CEPS est une structure administrative, incluant le
Ministère, les Caisses et l’UNCAM ; il négocie le prix avec
l’entreprise, négocie les volumes de ventes (au-delà des-
quels peuvent survenir des baisses de prix ou des remi-
ses). Ses décisions sont prises en fonction de l’ASMR, du
prix des comparateurs, des volumes de ventes, des prix
européens moyens. Le prix est publié par un avis au Jour-
nal Officiel.
Les délais moyens d’accès au marché, ajoutant les
démarches pour le prix et celles pour le remboursement,
sont très longs, d’environ 180
jours en France, qui se situe
parmi les pays les plus lents.
FIG. 2. —
Phases recherche/développement.
5 à 10 000
molécules
250
5
20 4 6 8 10 12 14
2 à 3
1
médicament
Années
Phase II
Phase III
DÉVELOPPEMENT
RECHERCHE
Dépôt dossier
& AMM
Identification
des cibles
Criblage
Pré-clinique
Phase I
Optimisation
prototype

I. Giri, F. Rouillon L’Encéphale, 2006 ;
32 :
858-60, cahier 3
S 860
Face à l’exigence croissante d’études post-AMM, la
place de chaque instance dans leur évaluation reste mal
définie : elle peut concerner la Commission de la Trans-
parence, la Direction Générale de la Santé ou l’Afssaps.
À l’Hôpital
Jusqu’à la récente réforme du Conseil de l’Hospitalisa-
tion, les prix de vente à l’Hôpital étaient libres, réglés par
des appels d’offres, et l’accès au médicament était immé-
diat après l’obtention de l’agrément des collectivités.
La création du Conseil de l’Hospitalisation a été conco-
mitante de la tarification à la pathologie. La situation est
désormais plus complexe : lorsque les médicaments sont
inclus dans les GHS (Groupes Homogènes de Séjour), la
procédure est la même qu’antérieurement, avec un prix
libre et un appel d’offres ; lorsque les médicaments sont
onéreux et hors des GHS (ce qui représente environ la
moitié des budgets hospitaliers de médicaments), le tarif
de responsabilité est alors négocié par le CEPS. C’est le
Conseil de l’Hospitalisation qui décide si les médicaments
doivent être classés dans les GHS ou en dehors. Il y a
donc une multiplication des étapes administratives.
Dans le prochain Projet de Loi sur le Financement de
La Sécurité Sociale (PLFSS), le CEPS sera aussi chargé
de contrôler les volumes de médicaments. Les prescrip-
tions devront désormais respecter les AMM ou certaines
indications validées soit par les référentiels officiels, soit
par des publications internationales.
Ces nouvelles procédures se traduisent par la signature
des Contrats de Bon Usage (CBU), signés entre l’hôpital
et l’Agence Régionale de l’Hospitalisation, prévoyant
molécule par molécule les volumes qui doivent être pres-
crits l’année suivante, avec un remboursement non plus
sur le budget global de l’hôpital mais par la Caisse d’Assu-
rance-maladie du patient.
Après la commercialisation
Les Laboratoires pharmaceutiques doivent encore,
avec les nouvelles lois sur l’assurance-maladie, prendre
en compte après la commercialisation un nouveau niveau
de régulation, au travers de la maîtrise médicalisée des
dépenses de santé : des engagements conventionnels ou
des accords de bon usage de soins sont signés entre
l’assurance-maladie et les syndicats de médecins, préci-
sant la manière dont les médicaments doivent être utilisés.
Ces différents avis (Afssaps, UNCAM, HAS) ne sont néan-
moins pas toujours cohérents entre eux…
Enfin, les assureurs complémentaires commencent à
jouer un rôle dans ces circuits complexes, en prenant
parfois en charge des produits refusés par l’assurance-
maladie, à la suite de leurs procédures propres d’éva-
luation.
1
/
3
100%