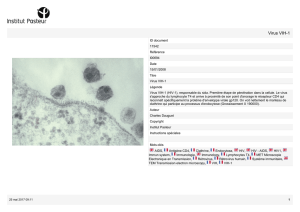
Analyse du profil d`expression de la protéine pro- apoptotique Dap

Analyse du profil d’expression de la protéine pro-
apoptotique Dap-3 dans les vésicules extracellulaires
produites dans le contexte de l’infection au VIH-1
Mémoire
Sofiane BERRAZOUANE
Maîtrise en microbiologie-immunologie
Maître ès sciences (M.Sc.)
Québec, Canada
© Sofiane BERRAZOUANE, 2016

Analyse du profil d’expression de la protéine pro-
apoptotique Dap-3 dans les vésicules extracellulaires
produites dans le contexte de l’infection au VIH-1
Mémoire
Sofiane BERRAZOUANE
Sous la direction de :
Caroline GILBERT, directrice de recherche

iii
Résumé
L’immunopathogenèse de l’infection au VIH-1 est principalement causée par la déplétion
des LT CD4 (lymphocytes T-CD4). Cette mort des LT CD4 dépend de plusieurs facteurs
comme la lyse des LT CD4 infectés et la présence de vésicules extracellulaires et
d’exosomes libérées par les cellules dendritiques et les LT CD4 infectés au VIH-1.
L’analyse protéomique des exosomes issus des cellules dendritiques mises en culture avec
le VIH-1 a révélé la présence de molécules pro-apoptotiques comme le Dap-3 (Death
Associated Protein 3). Nous avons proposé comme hypothèse que le Dap-3 puisse être
contenu dans d’autres types de vésicules extracellulaires et que le Dap-3 vésiculaire
contribue à la déplétion des LT CD4.
Après avoir optimisé l’immunobuvardage avec l’anti-Dap-3, nous avons déterminé la
présence de Dap-3 dans les vésicules extracellulaires issues des cellules RAJI-CD4-DCIR
infectées au VIH-1. L’utilisation de gradients de vélocité nous a permis d’observer la
présence de Dap-3 dans les fractions du gradient contenant les exosomes issus des cellules
RAJI-CD4-DCIR infectées, mais également dans d’autres fractions du gradient de vélocité
encore non caractérisées. Chez les patients, nous avons montré une hétérogénéité des
vésicules extracellulaires dans les fractions du gradient de vélocité issues des plasmas des
patients VIH-1+. Ces résultats indiquent la présence de plusieurs populations de vésicules
extracellulaires séparées par la méthode du gradient de vélocité. Enfin, la transfection des
cellules RAJI-CD4-DCIR et des cellules dendritiques a été mise au point avec les ARN
anti-sens de Dap-3 afin de produire éventuellement des vésicules Dap-3 négatives.
Ce projet de recherche aura permis de valider les outils nécessaires à la poursuite de l’étude
du rôle de Dap-3 dans la pathogenèse de l’infection au VIH-1.

iv
Table des matières
Résumé ........................................................................................... iii
Table des matières ......................................................................... iv
Liste des tableaux .......................................................................... vi
Liste des figures ........................................................................... viii
Liste des abréviations .................................................................... ix
Chapitre I : Introduction ............................................................... 1
1. Le virus de l’immunodéficience humaine de type 1 (VIH-1). 1
1.1. Découverte ....................................................................................................... 1
1.2. Épidémiologie du VIH-1 ................................................................................. 1
1.3. Structure du VIH-1 ......................................................................................... 1
1.4. Les phases d’infection au VIH-1 ................................................................... 2
1.4.1. Phase de primo-infection ..................................................................... 3
1.4.2. Phase chronique ............................................................................... 3
1.4.3. Phase du SIDA .................................................................................. 4
2. Les cellules participant à l’immunopathogenèse de l’infection
au VIH-1 .......................................................................................... 5
2.1. Les cellules dendritiques ................................................................................ 5
2.2. Les lymphocytes T CD4 .................................................................................. 7
2.3. Les lymphocytes T CD8 .................................................................................. 8
2.4. Déplétion des LT CD4 .................................................................................... 8
3. Apoptose ...................................................................................... 9
3.1. Apoptose des LT CD4 ................................................................................... 10
4. Les vésicules extracellulaires .................................................. 12
4.1. Découverte des exosomes ............................................................................. 12
4.2. Types de vésicules extracellulaires .................................................. 13
4.2.1. Les microvésicules .......................................................................... 13
4.2.2. Les vésicules apoptotiques ............................................................. 14
4.2.3. Les exosomes ................................................................................... 14

v
4.3. Les rôles pléiotropiques des vésicules extracellulaires dans la réponse
immunitaire .......................................................................................................... 16
4.4. Pathogenèse de l’infection au VIH-1 ......................................................... 17
4.5. Limitation des méthodes classiques ............................................................ 18
5. Mitochondries dans les infections au VIH-1 ......................................... 19
6. La protéine pro-apoptotique Dap-3 ....................................................... 20
6.1. Structure de Dap-3 ............................................................................ 21
6.2. Fonctions physiologiques de Dap-3 ................................................. 21
6.3. Rôles de Dap-3 dans les pathologies ................................................ 22
Chapitre II : Problématique, hypothèse et objectifs de recherche
........................................................................................................ 24
1. Problématique .......................................................................................... 24
2. Hypothèse de recherche .......................................................................... 24
3. Objectif ..................................................................................................... 25
Chapitre III : Matériel et Méthodes ........................................... 26
1. Patients ..................................................................................................... 26
2. Réactifs et anticorps ................................................................................ 26
2.1. Réactifs ............................................................................................... 26
2.2. Anticorps ............................................................................................ 27
3. Culture cellulaire ..................................................................................... 27
3.2.Cellules dendritiques ......................................................................... 28
4. Immunobuvardage .................................................................................. 28
5. Caractérisation des vésicules extracellulaires ...................................... 29
Purification des vésicules extracellulaires avec ultracentrifugation ... 29
Purification des vésicules extracellulaires par l’ExoQuick™ .............. 29
Séparation des vésicules extracellulaires par gradient de vélocité ...... 30
6. Détection des vésicules extracellulaires ................................................. 30
6.1. Mesure de l’activité d’acétylcholine estérase ................................. 30
6.2. Rayon d’hydrolyse dynamique des vésicules extracellulaires mesuré
au Zetasizer Nano ZS (Malvern™) ........................................................ 31
7. Transfection des cellules avec des ARN anti-sens anti-DAP3 ............. 31
8. Production virale ..................................................................................... 32
Chapitre IV : Résultats ................................................................ 33
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
1
/
83
100%