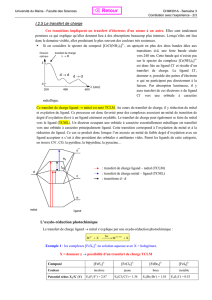
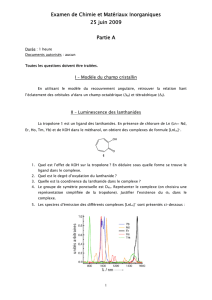
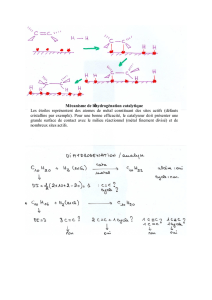
Coordination et réactivité des ligands

THÈSE
En vue de l’obtention du
DOCTORAT DE L’UNIVERSITÉ DE TOULOUSE
Délivré par l’Université Toulouse III - Paul Sabatier
Discipline : Chimie Spécialité : Physico-Chimie Théorique
Présentée et soutenue par Maxime MERCY
Le 4 Mars 2010
Coordination et Réactivité des ligands Bifonctionnels, une
étude théorique
JURY
Stuart A. Macgregor
Professeur à l’Université Heriot Watt, Edinburgh, Royaume-Uni (Rapporteur)
Jeremy Harvey
Professeur à l’Université de Bristol, Royaume-Uni (Rapporteur)
Jean-François Carpentier
Professeur à l’Université Rennes I, France (Examinateur)
Karinne Miqueu
Chargé de recherche à l’Université de Pau et des Pays de l’Adour, France (Examinatrice)
Blanca Martin-Vaca
Professeur à l’Université Toulouse III, France (Examinatrice)
Laurent Maron
Professeur à l’Université Toulouse III, France (Directeur de Thèse)
Ecole doctorale : Sciences de la matière
Unité de recherche : LPCNO (UMR 5215)


Remerciements
Cette thèse représente trois années de ma vie durant lesquelles j’ai côtoyé de nom-
breuses personnes qui ont toutes plus ou moins contribué à ce manuscrit. Dans cette
première page, je tiens à remercier toutes les personnes, chercheurs, enseignants, étu-
diants, familles, amis, qui m’ont permis d’arriver si loin.
Cette thèse s’est déroulée au LPCNO dans l’équipe modélisation. Je remercie tout
le laboratoire et en particulier l’équipe, Romuald, Franck, Lionel, Iann, qui m’ont donné
les moyens d’effectuer cette thèse et qui n’ont pas été avare en discussion. Entre autre,
merci Franck pour la gestion des clusters, c’est grâce à toi que j’ai pu effectuer des
milliers d’heures de calculs et merci Romuald pour toute les astuces logiciels. En parti-
culier, je remercie dans l’équipe Laurent Maron, mon chef, qui m’a fait confiance pour
travailler avec lui sur l’ANR Bili. Tu as réussi dans ton encadrement à nous laisser une
certaine liberté et indépendance tout en nous guidant avec une main parfois ferme. En
plus, tu n’as pas été avare sur les frais (cool les ordis, les conférences).
Ce travail théorique s’accompagne d’un impressionnant travail expérimental. Ainsi,
merci à l’équipe de Didier Bourissou du LHFA pour cette collaboration très productive.
Ce travail commun entre expérience et théorie grâce à ces étranges ligands ambiphiles
est une nouvelle pierre à la transversalité entre le monde des processeurs et celui des
éprouvettes. Merci Ghenwa, Abderrahmane, Pauline, et un merci spéciale à Marie Sir-
coglou, mon alter-ego thésard expérimentateur sur ce sujet de thèse.
Enfin merci à toutes les personnes que j’ai pu rencontrer durant mon cursus à
l’université et qui m’ont encadré avec beaucoup de bienveillance. Notamment les en-
seignants de l’option chimie théorique. Je n’oublie pas mes camarades,Marie-Laure,
Iker, Benoit, Jean-Pierre. Nous avons bien travaillé ensemble et nous nous sommes
bien amusés durant notre cursus. Ensuite Noemi, qui m’a guidé dans mes premiers
calculs sur les éléments f, et sur la réactivité. Tu as su nous apprendre beaucoup de
choses d’un point de vue scientifique, mais aussi sur les rouages d’un système parfois
sombre et obscure (de l’université et du CNRS). Bien entendu, je n’oublie pas le club
Mickey, son président Iker et ses membres Léa, Ahmed, Nicolas, Ludo, avec qui les
soirées loups-garous, les discussions et les bouffes ont créé une ambiance de travaille
très très agréable. Un merci très particulier pour Guillaume, merci d’avoir partager ces
3 années avec moi. Bien évidemment, mes pensées vont aussi vers toute ma famille


La science consiste à passer d’un étonnement à un autre.
Aristote
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
192
193
194
195
196
197
198
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
192
193
194
195
196
197
198
1
/
198
100%