Stage - DLST

Université Joseph Fourier
Département Licence Sciences & Technologies
RAPPORT DE STAGE
Synthèse et Utilisation d'Organocatalyseurs Chiraux à l'Iode
Hypervalent
Mahamadou MAIGA
Laboratoire d’accueil : Département de Chimie Moléculaire (DCM)
Directeur du laboratoire : Serge COSNIER
Responsable du stage : Dr Florian BERTHIOL
Licence Chimie-Biologie 1ère Année
Année Universitaire : 2014-2015

Remerciements
Tout d’abord, mes sincères remerciements sont destinés au directeur du département de
chimie moléculaire (DCM), Serge COSNIER qui en répondant à ma demande m’a permis
d’effectuer ce stage au sein du DCM dans l’équipe Synthèse Et Réactivité en Chimie Organique
(SERCO) afin que je puisse découvrir le monde de la recherche et améliorer mon niveau de travaux
pratiques en chimie.
Je remercie également mon maître de stage le Dr Florian BERTHIOL pour son encadrement, ses
explications et enseignement qu’il m’a apporté tout au long de ce stage. De plus, chacun de ses
conseils, remarques ou explications ont contribué à mon enrichissement durant ce mois. Je lui suis
très reconnaissant pour la confiance qu'il m’a accordé et je le remercie pour son entière
disponibilité.
Pour finir je remercie les membres de l’équipe SERCO pour leur accueil et toute leur
bienveillance favorisant le bon déroulement de mon stage.

SOMMAIRE :
I) Introduction ………………………………………………………....1
II) Présentation de la structure d'accueil …………………………….1
III) Synthèse des Organocatalyseurs
1) Méthodes de Synthèse du 2,2'-(2-iodobenzène-1,3-diyl)bis(4,5-dihydro-
1,3-oxazole)
a) Prémière méthode …………………………………………………...3
b) Séconde méthode …………………………………………………....4
c) Troisième méthode …………………………………………………..4
2) Synthèse du catalyseur oxide de N-[(Z)-(2-iodophényl)méthylidène]-
N- (1-phényléthyl)amine …………………………………………………..4
3) Synthèse du catalyseur de type bis nitrone …………………………….5
4) Synthèse du catalyseur oxide de N-[(Z)-(2-iodo-3-
méthylphényl)méthylidène]-N-[(1R)-1-phényléthyl]amine
a) Synthèse de l'acétate de (2-iodo-3-méthyl)benzyle…………………5
b) Synthèse du (2-iodo-3-méthylphényl)méthanol ………………………5
c) Synthèse du (2-iodo-3-méthylphényl)méthanal ………………………6
d) Synthèse de l'oxide de N- [(Z)-(2-iodo-3-méthylphényl)méthylidène]-
N-[(1R)-1- phényléthyl]amine……………………………………..6
IV) Utilisation des Organocatalyseurs …………………………………………...6
Conclusion …………………………………………………………………………7
Généralité : Principe des Techniques Utilisées …………...……………………...8
Protocoles expérimentales ………………………….……………………………..9
Bibliographie ………………………………………….…………………………...13
0

I) Introduction:
Avec comme ambition des études dans le domaine de la chimie je cherchais un stage dans
ce domaine-là. J’ai été très passionné par les cours de chimie organique dispensés au cours de ce
semestre. J’ai alors cherché à effectuer mon stage d’excellence dans ce domaine. C'est ainsi que j'ai
postulé pour la proposition de stage du Dr Berthiol qui m'intéressait beaucoup et il a accepté de me
prendre en stage.
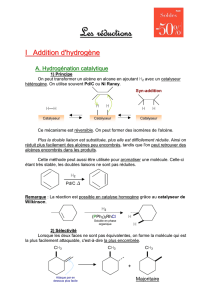
Durant ce mois, mon projet fut de synthétiser et utiliser des catalyseurs bis-nitrone, bis-
oxazoline tous à l'iode hypervalent (voir Annexe 1). Après leurs synthèse, ces catalyseurs seront
utilisés dans des réactions d'oxytosylation de cétones pour tester leur efficacité.
L'un des objectifs lors de cette synthèse était de me montrer les différentes méthodes
classiquement utilisées en chimie organique mais également de synthétiser des catalyseurs qui
serviront dans des réactions d'oxytosylation. L'utilisation de ces organocatalyseurs vont permettre
d'estimer leur efficacité grâce aux rendements et aux excès énantiomériques (ee) qui seront obtenus
en oxydation. On souhaite atteindre avec ces nouveaux catalyseurs les meilleurs rendements et les
meilleurs excès énantiomériques possibles.
Quant à l’organisation de mon rapport, dans un premier temps je présente la structure
d’accueil où a eu lieu ce stage, puis j’ai détaillé le déroulement de celui-ci en ce qui concerne le
travail que j’ai réalisé et ce que j'ai appris durant ce mois.
II) Présentation de la structure d'accueil:
Le Département de Chimie Moléculaire (DCM) de Grenoble est une unité mixte de
recherche (UMR-5250) associant le CNRS et l’Université Joseph Fourier à Grenoble. Créé le 01
Janvier 2007 par le regroupement des unités LEDSS (UMR-5616) et LEOPR (UMR-5630), le
DCM mobilise 150 personnes autour de deux axes de recherches interactifs qui sont la chimie pour
la santé et la chimie pour les nanosciences.
Il est structuré en 5 grandes thématiques scientifiques dont l’organisation, l’animation et
l’articulation administrative, scientifique et technique sont assurées par un coordonnateur. En effet
ces équipes de recherche sont:
•SERCO Synthèse Et Réactivité en Chimie Organique
•CIRE Chimie Inorganique REdox
1

•I2BM Ingénierie et Interactions BioMoléculaires
•BEA Biosystèmes Electrochimiques et Analytiques
•CT Chimie Théorique
Ces équipes de recherches disposent d’un solide soutient technique de :
•ICMG Institut de Chimie Moléculaire de Grenoble
•Plateforme Physico-Chimie
•Plateforme Synthèse
•Informatique
L’unité accueille environ 26 doctorants et 15 postdoctorants. 8 thèses par an sont soutenues
en moyenne. Le DCM bénéficie de personnel, d’équipements et de technologies du meilleur niveau
en soutien des recherches (RMN, RPE, cristallographie, spectrométrie de masse, microanalyse,
serveur de calcul et grappe PC, AFM, SPR, QCM, spectroscopie UV, stopped-flow etc....).
L’unité est impliquée dans de nombreux programmes régionaux, nationaux et internationaux. Elle
est également étroitement associée à des actions tournées vers des applications thérapeutiques,
diagnostiques et technologiques en partenariat avec des laboratoires universitaires et industriels.
Au cours de mon stage j'ai travaillé au sein de l'équipe de SERCO qui regroupe les activités
du DCM dans le domaine des nouvelles méthodes de synthèse en chimie organique et des nouvelles
stratégies pour la préparation de produits naturels complexes ou de molécules biologiquement
actives. Elle est spécialisée dans les domaines suivants:
•Synthèse de Produits Naturels
•Nouvelles Réactivités en Synthèse
•Synthèse, Réactivité et Valorisation de Nouveaux Composés Hétérocycliques
•Catalyse et Méthodologies de Synthèse
Voir l'organigramme du DCM en annexe 2.
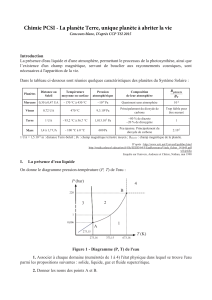
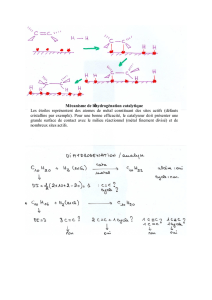
Déroulement Du Stage:Travaux réalisés et connaissances acquises :
Ce stage m’a permis de découvrir les différentes étapes de ce projet de recherche,
allant de la synthèse d'organocatalyseurs chiraux à l'iode hypervalent jusqu’à leur utilisation dans
les réactions d'α-oxytosylation de cétones.1,2 C'est pourquoi dans un premier temps je vais axer mon
étude sur la synthèse de ces catalyseurs et enfin sur leurs utilisations dans les réactions d' α-
2
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
1
/
20
100%