réaction électrode

Electrochimie et applications 179
C. LA SURTENSION D’ACTIVATION OU DE TRANSFERT DE CHARGES
1. Introduction
On parlera de surtension d’activation ou de transfert de charges dans le cas où la vitesse
globale de la réaction est limitée par le transfert d’électrons.
Le transfert de charges sera fonction du potentiel et de la nature du métal.
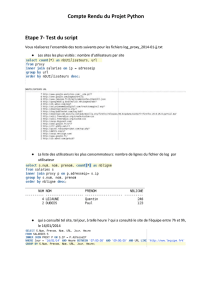
On distingue, du point de vue pratique, 3 types de systèmes :
1°) les systèmes réversibles
2°) les systèmes partiellement irréversibles
3°) les systèmes irréversibles (ou complètement polarisables)
Figure VIII.17. : Courant d’échange.
Pour étudier uniquement le transfert de charges et ne pas être ennuyé par le phénomène
de diffusion, on agite la solution (diminution de l'épaisseur de la couche de diffusion) et
on travaille avec un électrolyte concentré.
Butler - Volmer
-1,0E+01
-8,0E+00
-6,0E+00
-4,0E+00
-2,0E+00
0,0E+00
2,0E+00
4,0E+00
6,0E+00
8,0E+00
1,0E+01
-0,6 -0,4 -0,2 0 0,2 0,4 0,6
V
i (A/cm2)
1
1,00E-03
1,00E-08
io =
Eéq
réversible
réversible
irréversible
i01 > i02 > i03
i0=

Electrochimie et applications 180
2. Importance de la surtension d’activation (de la limitation du transfert de charges)
Exemples :
Le dépôt des métaux moins nobles que l’hydrogène est possible, même en solution
acide lorsque la surtension de dégagement d’hydrogène H2 est élevée.
Le système Cd/Cd2+ possède un E°Cd/Cd2+ = - 0,4 V/ENH
En milieu acide, la thermodynamique nous donne :
E°Cd/Cd2+ = - 0,4 V/ENH
E°H2/H+ = 0 V/ENH
La cinétique nous indique que la surtension de dépôt du cadmium est faible, tandis
que la surtension de dégagement d'hydrogène sur cadmium vaut - 1 V.
En résumé :
Cd2+/Cd = 0,1 à 0,2 V
H2/Cd = 1 V
Malgré les prévisions de la thermodynamique, on pourra déposer tout le cadmium
en milieu acide.
2
/
0CdCd
E
HH
E/
0
2
CdeCd 2
2
i(mA/dm2)
E(V/ER)
CdH/
2
2
/
0CdCd
E
HH
E/
0
2
CdeCd 2
2
i(mA/dm2)
E(V/ER)
CdH/
2
Figure VIII.18 : Surtension de dégagement d’hydrogène sur cadmium.
Cas du mercure
La surtension de dégagement d'H2 sur mercure est très élevée, on peut effectuer
des réductions sur électrodes de Hg avant le dégagement d'H2.
Différentes applications pratiques sont basées sur ce principe (Ex : polarographie,
électrolyse du NaCl, étude de la double couche électrochimique).

Electrochimie et applications 181
Si on veut faire fonctionner une pile à combustible H2-O2, il faut que la combustion de
l'H2 (oxydation anodique) et la réduction de l'oxygène (réduction cathodique) se passent
le plus facilement possible pour limiter les pertes.
Il est donc nécessaire dans ce cas d'utiliser comme matériaux d'électrodes des métaux
ou matériaux catalytiques qui limitent les surtensions correspondantes.
3. Surtension de décharge ou d’activation - Etablissement de la relation i = f()
a) Calcul de la vitesse d'une réaction chimique - Rappel
Rappel de la théorie du complexe activé.
Au cours d'une réaction chimique élémentaire :
A + B C + D
L'énergie potentielle du système évolue d'un minimum local correspondant à l'état de
départ vers un autre minimum local correspondant au produit final.
Entre l'état initial et l'état final, l'énergie potentielle du système passe par une valeur
maximale correspondant à une configuration particulière du système appelée complexe
activé notée x*.
On a représenté à la figure VIII.19., les variations d'énergie du système au cours de la
réaction.
Figure VIII.19. : Variations d’énergie lors d’une oxydation.
La théorie du complexe activé conduit à une expression de la vitesse de la forme :

Electrochimie et applications 182
V = k . A . B
où k, constante de vitesse de réaction est donnée par la loi d'Arrhénius :
k = P . exp
RT
*
G
(p constante)
G* est la différence d'énergie entre le complexe activé et les réactifs, on l'appelle
également énergie d'activation. La valeur de l'énergie d'activation peut fournir des
indications sur le régime cinétique de la réaction :
G* est de l'ordre de 2 à 5 kcal/mole en régime de diffusion et de l'ordre de 10 à 20
kcal/mole en régime de réaction.
b) Calcul des vitesses de transfert de charge
De même, le passage d'une espèce chargée à travers la double couche implique le
franchissement d'une barrière d'énergie dont la hauteur dépend de la différence d'énergie
entre l'état initial et l'état activé.
Soit la réaction :
ionisation
Me Men+ + ne
décharge
Considérons cette réaction comme formée par deux actes antagonistes dont les vitesses
respectives sont
v
et
v
A tout moment, la vitesse globale est :
v = v - v
Et pour les réactions électrochimiques,
( . s + ne Produits), > 0 pour les oxydants
i =
nF
. V =
nF
[ S] . k 0 . e (*)
Il en résulte que si on pose :
i+ = n F
v
et i - = n F
v
On tire : i = i+ - i-
Eact
RT

Electrochimie et applications 183
Me SOLUTION
i+
Si E > Eeq > 0 et i > 0 i-
courant anodique
Si E = Eeq = 0 et
i = i + - i- = 0 i+
i + = i- = i0 i-
i0 courant d'échange
Si E < Eeq < 0 et i < 0 i+
courant cathodique i-
c) Hypothèses pour le calcul de la surtension d'activation
a) On utilise le concept d'activation et la constante k dérive de la loi d'Arrhénius
(comme pour une réaction chimique).
k k e
oERT
act
./
b) Le transfert de charges s'effectue à une distance finie de l'électrode au sein de la
double couche compacte où le potentiel peut être différent de celui de l'électrode et de
celui du coeur de la solution (figure VIII.20.).
Si = 0 le potentiel du coeur de la solution est identique à celui de la limite de la
couche compacte (cas des solutions électrolytiques concentrées J > 0,02).
Figure VIII.20. : Influence de la force ionique J sur l’épaisseur de la double couche.
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
1
/
22
100%