Lire l`article complet

Correspondances en Métabolismes Hormones Diabètes et Nutrition - Vol. XVI - nos 1-2 - janvier-février 2012
32
dossier thématique
Conséquences endocriniennes
et métaboliques du stress
Les glucocorticoïdes inhibent
le développement des cellules β
pancréatiques. Une étape importante
dans la pathogenèse du diabète de type 2 ?
Glucocorticoids inhibit pancreatic β-cell development.
A crucial step in the pathogenesis of type 2 diabetes?
Bérangère Valtat* , Bertrand Blondeau*
* Centre de recherche des
Cordeliers, Inserm U872,
équipe 8, université
Paris-VI, Paris.
Points forts
Highlights
»La fonction et la masse des cellules β pancréatiques jouent un
rôle crucial dans la progression du diabète de type 2.
»
Un environnement intra-utérin anormal programme les altérations
de la masse et de la fonction sécrétrice des cellules β adultes.
»
Les hormones glucocorticoïdes sont de puissants inhibiteurs
de la fonction, mais aussi du développement des cellules β
pancréatiques.
Mots-clés : Diabète de type 2 − Cellules β − Glucocorticoïdes −
Programmation fœtale.
Pancreatic beta-cell function and mass play a crucial role in
the progression of type 2 diabetes.
Abnormal intra-uterine environment programs the alterations
of adult beta-cell mass and secretory function.
Glucocorticoid hormones are potent inhibitors of both beta-
cell function and development.
Keywords: Type 2 diabetes − β cells − Glucocorticoids − Fetal
programming.
L
e diabète de type 2 (DT2) est et sera une patho-
logie chronique majeure du xxie siècle. Durant
les dernières décennies, la prévalence globale du
DT2 dans la population mondiale a augmenté selon des
proportions épidémiques. Actuellement, le DT2 touche
plus de 280 millions de personnes dans le monde, et ce
chiffre pourrait atteindre 438 millions en 2030.
Cette maladie métabolique se caractérise par une hyper-
glycémie chronique, due à une altération de la sécrétion
d’insuline par les cellules β du pancréas et à une résistance
à l’insuline des tissus périphériques (le foie, le muscle et
le tissu adipeux). Plusieurs facteurs contribuent à l’appa-
rition du DT2 : une prédisposition génétique et l’influence
de facteurs environnementaux (surcharge pondérale,
sédentarité). Il est maintenant clairement admis que la
transition de l’état prédiabétique au diabète franc repose
sur un épuisement des cellules β du pancréas, qui sont
incapables de faire face à la pression de la résistance à
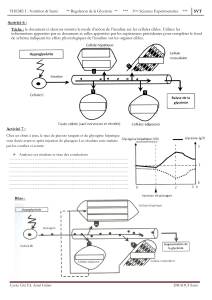
l’insuline (figure 1, d’après [1]). L’insuline, sécrétée par
les cellules β, est la seule hormone hypoglycémiante de
l’organisme. La normoglycémie est maintenue par un
équilibre entre l’action et la sécrétion de l’insuline. On a
longtemps pensé que seule la sécrétion d’insuline et ses
anomalies participaient à la mise en place du DT2, mais
de récentes données suggèrent que le nombre de cellules
β présentes dans le pancréas (masse de cellules β) joue
aussi un rôle important dans la progression de la maladie.
Dysfonction de la cellule β : rôle central
dans la progression du diabète de type 2
La cellule β pancréatique module sa fonction sécrétrice
en réponse à des changements de l’action de l’insuline
sur ses tissus cibles (principalement foie, muscle, tissu
adipeux). Pour évaluer la fonction de la cellule β, une
technique couramment utilisée est le “disposition index”
(figure 2, d’après [2]). C’est une relation curviligne qui
illustre la variation de la sécrétion d’insuline par la cellule β
en fonction du degré de sensibilité à l’insuline des tissus
périphériques. Pour une sensibilité à l’insuline équivalente,
un individu normoglycémique deviendra intolérant au
glucose, voire diabétique de type 2, si sa sécrétion d’insu-

Correspondances en Métabolismes Hormones Diabètes et Nutrition - Vol. XVI - nos 1-2 - janvier-février 2012
33
Les glucocorticoïdes inhibent le développement des cellules β pancréatiques.
Une étape importante dans la pathogenèse du diabète de type 2 ?
line n’est pas suffisante (3). Dans les phases précoces du
DT2, la résistance à l’insuline est donc compensée par
une augmentation de la sécrétion d’insuline. Des études
fonctionnelles montrent cependant que les individus qui
sécrètent peu d’insuline en réponse à une charge orale en
glucose ont un risque accru de développer ultérieurement
un DT2. À l’inverse, la résistance à l’insuline évolue peu
durant la progression de l’intolérance glucidique au dia-
bète manifeste, alors que la fonction de la cellule β subit
d’importants changements (4). Le défaut de la fonction
de la cellule β serait donc à l’origine de la transition de
l’intolérance au glucose au développement d’un DT2. Ces
résultats, reproduits dans l’étude UKPDS (United Kingdom
Prospective Diabetes Study), qui a suivi plus de 5 000 per-
sonnes sur 20 ans, ont permis de proposer le concept
selon lequel la dysfonction de la cellule β ne se produirait
pas seulement lors de la transition de la normotolérance
à l’intolérance au glucose, mais serait en fait l’événement
pathogénique clé induisant cette progression (figure 1) [5].
Diminution de la masse de cellules β : unrôle
dans la pathogenèse du diabète de type 2 ?
Alors que le rôle de la dysfonction de la cellule β est
clairement démontré dans le développement du DT2,
l’importance de la diminution de la masse de cellules β
reste controversée.
De récentes études ont démontré que la masse de cel-
lules β est diminuée chez les diabétiques de type 2.
En particulier, l’équipe de P. Butler a montré, à partir
d’autopsies pancréatiques, que les sujets avec une
intolérance au glucose et un DT2 présentaient une
réduction de la fraction de cellules β par rapport aux
sujets normotolérants au glucose (6).
Il semble donc que la régulation de l’homéostasie gluci-
dique chez l’adulte dépende à la fois d’un nombre adéquat
de cellules β, de leur plasticité au cours de la vie et de
leur fonction sécrétrice en réponse au glucose. Le déve-
loppement des cellules β est une étape cruciale pour la
mise en place de ces paramètres. Des modifications de
l’environnement intra-utérin affectant ce développement
peuvent avoir des conséquences délétères sur l’homéos-
tasie glucidique à l’âge adulte. Il est donc important de
comprendre les mécanismes impliqués dans le dévelop-
pement des cellules β et comment des altérations au cours
du développement entraînent un DT2 à l’âge adulte (7).
La programmation fœtale de certaines
pathologies adultes
Au début des années 1990, C.N. Hales et D.J. Barker ont
suggéré que des altérations de l’environnement fœtal
pouvaient être à l’origine de certains désordres cardio-
vasculaires et métaboliques chez l’adulte (8). La notion
de programmation fœtale des maladies adultes a été
renforcée par de nombreuses études épidémiologiques,
associant la sous-nutrition durant la gestation, le retard
de croissance intra-utérin (RCIU) caractérisé par un petit
poids à la naissance, et le risque accru de développer
une maladie métabolique telle que le DT2 à l’âge adulte.
Figure 1. Modèle actuel de mise en place du DT2, incluant la résistance à l’insuline, le dysfonc-
tionnement des cellules β et la réduction de masse des cellules β
(d’après [1])
.
Résistance à l’insuline
Défauts acquis
Prédiabète Diabète de type 2
Tolérance au
glucose normale
Dysfonction des cellules β Épuisement
des cellules β
Masse des cellules β
diminuée de 40 %
Production hépatique
de glucose augmentée
Masse des cellules β
diminuée de 40 à 80 %
Figure 2. Représentation schématique du disposition index, c’est-à-dire de la relation entre la
fonction de la cellule β et la sensibilité à l’insuline (2). Pour une personne normotolérante, il
existe une relation hyperbolique entre la fonction de la cellule β et la sensibilité à l’insuline. Des
déviations de cette hyperbole correspondent au développement d’une intolérance au glucose,
puis d’un diabète de type 2 (DT2).
Résistance à l’insuline avec compensation de la cellule β
Résistance à l’insuline sans compensation de la cellule β
Normotolérant
Intolérant au glucoseDT2
Sensibilité à l’insuline
Fonction de la cellule β

Correspondances en Métabolismes Hormones Diabètes et Nutrition - Vol. XVI - nos 1-2 - janvier-février 2012
34
dossier thématique
Conséquences endocriniennes
et métaboliques du stress
Chez l’homme, l’étude des mécanismes impliqués dans
le RCIU et des altérations primitives du développement
embryonnaire, et plus spécifiquement du pancréas
endocrine, se révèle difficile. De nombreux modèles
animaux ont donc été étudiés pour comprendre les
relations physiologiques et moléculaires entre le RCIU
provenant de l’environnement maternel et le DT2. En
particulier, le modèle de restriction de l’apport calorique
par sous-nutrition maternelle développé au labora-
toire a permis de mettre en avant les facteurs et les
mécanismes impliqués dans l’association entre l’envi-
ronnement intra-utérin et le DT2, et notamment l’effet
délétère de la surexposition fœtale aux glucocorticoïdes
(GC) sur le développement des cellules β.
Rôle physiologique des glucocorticoïdes
au cours de la vie fœtale
Les GC sont des hormones pléiotropes impliquées entre
autres dans la régulation du comportement, du système
immunitaire et de l’homéostasie glucidique. Ils jouent
également un rôle primordial dans le développement
normal du fœtus et la maturation des organes fœtaux.
Chez l’homme comme chez de nombreuses espèces
animales, l’augmentation des concentrations de cortisol
en fin de gestation coïncide avec la maturation des
organes fœtaux (poumons, thymus, tractus digestif,
etc.) [9].
Au cours de la grossesse, les GC exercent un effet béné-
fique sur la maturation des organes fœtaux, mais ils
peuvent aussi avoir des effets délétères sur la croissance
intra-utérine et le développement des cellules β quand
ils sont présents à des concentrations excessives.
Rôle des glucocorticoïdes dans le modèle
de restriction de l’apport calorique
Pour étudier les relations entre une modification de l’en-
vironnement fœtal et l’apparition du DT2, le laboratoire
a développé un modèle de sous-nutrition maternelle
chez le rat correspondant à une restriction calorique
globale (50 % de la ration alimentaire quotidienne)
pendant la dernière semaine de gestation. Les nouveau-
nés issus des rates malnourries présentent un RCIU
caractérisé par un petit poids de naissance (10). Les
travaux de l’équipe ont montré qu’au cours de la période
fœtale de sous-nutrition maternelle, la corticostéroné-
mie maternelle et fœtale était anormalement élevée en
fin de gestation et que la masse de cellules β chez les
fœtus malnourris était diminuée (11). Des approches
à la fois chirurgicales et pharmacologiques nous ont
permis de définir les GC comme des régulateurs néga-
tifs du développement des cellules β en situation de
sous-nutrition ou de nutrition normale.
Conséquences métaboliques
de la surexposition in utero
aux glucocorticoïdes
Chez de nombreuses espèces animales, le traitement
in utero aux GC conduit à un RCIU (12). D’autres études
ont pu mettre en évidence le lien entre la surexposition
in utero aux GC et l’apparition de troubles métabo-
liques à l’âge adulte. Des rats surexposés artificiel-
lement aux GC durant leur vie fœtale ont un poids
de naissance fortement réduit et des contenus pan-
créatiques en insuline diminués. À l’âge adulte, les
fœtus ainsi exposés présentent une hypertension, une
hyperglycémie, une hyperinsulinémie et une résistance
à l’insuline (13).
Modèle d’inactivation du récepteur
aux glucocorticoïdes dans les cellules β
Le modèle de sous-nutrition fœtale décrit précédem-
ment a permis de montrer que les GC jouaient un rôle
majeur dans le contrôle du développement des cellules
β. Pour définir de façon précise le mécanisme d’action
des GC sur leur développement, nous avons étudié les
conséquences de l’absence de signalisation GC sur ce
paramètre en invalidant spécifiquement le récepteur
aux GC (GR) dans les cellules pancréatiques précurseurs
(souris GRPrec). Les souris GRPrec présentent 2 fois plus
de cellules β que les animaux contrôles, ainsi qu'un
nombre d’îlots augmenté (14). Cela suggère que, lorsque
la voie inhibitrice des GC est rendue inactive, les précur-
seurs du pancréas se différencient préférentiellement
en cellules β. Néanmoins, l’étude de fœtus de souris
invalidées pour le GR dans tout l’organisme (GRnull/null)
a permis de montrer que les premières étapes de la
mise en place du pancréas et des précurseurs, qui vont
devenir des cellules β, ne dépendaient pas de la voie de
signalisation des GC (15). Seules les étapes plus tardives
du développement pancréatique, comme le choix des
précurseurs de se différencier ou non en cellules β, sont
finement régulées par les GC.
Le récepteur aux glucocorticoïdes
estnécessaire aux effets délétères
de la sous-nutrition maternelle
sur les cellules β
Pour mettre en évidence l’effet direct de la voie de
signalisation des GC sur le développement des cellules

Les glucocorticoïdes inhibent le développement des cellules β pancréatiques.
Une étape importante dans la pathogenèse du diabète de type 2 ?
β en condition de surexposition fœtale aux GC, nous
avons donc transposé notre modèle de sous-nutrition
maternelle des rates gestantes aux souris, afin de pou-
voir étudier les modifications de l’environnement fœtal
sur nos souris mutantes invalidées pour le GR. Cette
étude a permis de distinguer les effets de la voie de
signalisation des GC passant par l’activation du GR des
effets résultant de facteurs modifiés par l’état nutri-
tionnel de la mère durant la gestation. Nous avons
retrouvé chez la souris les conséquences délétères de
la sous-nutrition maternelle : la restriction calorique
chez la femelle gestante induit une hypoglycémie et
une hypercorticostéronémie maternelles et fœtales.
Ces modifications engendrent dans la descendance un
RCIU, une diminution permanente et irréversible de la
masse de cellules β et une intolérance au glucose avec
une altération de la sécrétion d’insuline en réponse au
glucose chez l’adulte. Lors de la surexposition fœtale aux
GC, l’absence de GR dans les précurseurs pancréatiques
chez les souris GRPrec empêche la régulation négative
des GC sur le développement des cellules β (16), ce qui
atteste clairement que les effets délétères de la sous-
nutrition maternelle sur le développement des cellules β
au cours de la gestation sont dépendants de la présence
du GR dans les cellules précurseurs pancréatiques.
En conclusion, l’ensemble des données présentées ici
montre que des altérations de l’environnement intra-
utérin dans lequel se développe le fœtus augmentent
la susceptibilité à développer un DT2, induit par une
altération des cellules β. Nous avons explicitement mis
en évidence le fait que la surexposition fœtale aux GC
altérait le développement des cellules β pancréatiques,
conduisant à un nombre réduit de cellules β qui pré-
sentent également un défaut de sécrétion d’insuline
(figure 3). Ces résultats, transposés chez l’homme, sug-
gèrent que le stress prénatal, potentielle source de
surexposition des fœtus aux GC, peut être un élément
étiologique majeur dans les altérations de la masse et
de la fonction des cellules β. ■
Figure 3. La surexposition fœtale aux glucocorticoïdes (GC) affecte le développement des
cellules β, avec des répercussions à la fois sur leur fonction et sur leur nombre, augmentant ainsi
le risque de développer à l’âge adulte un DT2. Les conséquences des modifications intra-utérines
sur les tissus sensibles à l’insuline (foie, muscle, tissu adipeux) participent également à cette
maladie, mais elles ne sont pas décrites ici.
Modifications de l’environnement
intra-utérin
Diabète de type 2
Insulinorésistance
+
défaut de masse et de fonction β
Cellules β
Tissus
insulinosensibles
1. Leahy JL. Pathogenesis of type 2 diabetes mellitus. Arch
Med Res 2005;36(3):197-209.
2. Stumvoll M, Goldstein BJ, van Haeften TW. Type 2 dia-
betes: principles of pathogenesis and therapy. Lancet
2005;365(9467):1333-46.
3. Kahn SE, Prigeon RL, McCulloch DK et al. Quantification
of the relationship between insulin sensitivity and beta-cell
function in human subjects. Evidence for a hyperbolic function.
Diabetes 1993;42(11): 1663-72.
4.
Weyer C, Bogardus C, Mott DM, Pratley RE. The natural his-
tory of insulin secretory dysfunction and insulin resistance
in the pathogenesis of type 2 diabetes mellitus. J Clin Invest
1999;104(6):787-94.
5.
Turner RC, Cull CA, Frighi V, Holman RR. Glycemic control
with diet, sulfonylurea, metformin, or insulin in patients with
type 2 diabetes mellitus: progressive requirement for multiple
therapies (UKPDS 49). UK Prospective Diabetes Study (UKPDS)
Group. JAMA 1999;281(21):2005-12.
6. Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA,
Butler PC. Beta-cell deficit and increased beta-cell apoptosis
in humans with type 2 diabetes. Diabetes 2003;52(1):102-10.
7. Bréant B, Gesina E, Blondeau B. Nutrition, glucocorticoids
and pancreas development. Horm Res 2006;65(Suppl.3):98-104.
8. Barker DJ, Hales CN, Fall CH, Osmond C, Phipps K, Clark PM.
Type 2 (non-insulin-dependent) diabetes mellitus, hypertension
and hyperlipidaemia (syndrome X): relation to reduced fetal
growth. Diabetologia 1993;36(1):62-7.
9. Smith ID, Shearman RP. Fetal plasma steroids in relation
to parturition. I. The effect of gestational age upon umbilical
plasma corticosteroid levels following vaginal delivery. J Obstet
Gynaecol Br Commonw 1974;81(1):11-5.
10. Garofano A, Czernichow P, Bréant B. In utero under-
nutrition impairs rat beta-cell development. Diabetologia
1997;40(10):1231-4.
11. Blondeau B, Lesage J, Czernichow P, Dupouy JP, Bréant B.
Glucocorticoids impair fetal beta-cell development in rats. Am
J Physiol Endocrinol Metab 2001;281(3):E592-9.
12. Reinisch JM, Simon NG, Karow WG, Gandelman R. Prenatal
exposure to prednisone in humans and animals retards intra-
uterine growth. Science 1978;202(4366):436-8.
13.
Nyirenda MJ, Seckl JR. Intrauterine events and the pro-
gramming of adulthood disease: the role of fetal glucocorticoid
exposure (Review). Int J Mol Med 1998;2(5):607-14.
14.
Gesina E, Tronche F, Herrera P et al. Dissecting the role
of glucocorticoids on pancreas development. Diabetes
2004;53(9):2322-9.
15.
Gesina E, Blondeau B, Milet A et al. Glucocorticoid signal-
ling affects pancreatic development through both direct and
indirect effects. Diabetologia 2006;49(12):2939-47.
16.
Valtat B, Dupuis C, Zenaty D et al. Genetic evidence of the
programming of beta cell mass and function by glucocorticoids
in mice. Diabetologia 2011;54(2):350-9.
Références
1
/
4
100%