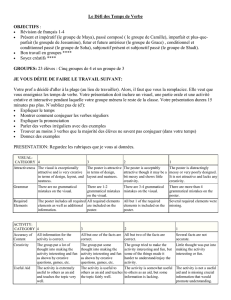
EP 0888345 B1

Note: Within nine months of the publication of the mention of the grant of the European patent in the European Patent
Bulletin, any person may give notice to the European Patent Office of opposition to that patent, in accordance with the
Implementing Regulations. Notice of opposition shall not be deemed to have been filed until the opposition fee has been
paid. (Art. 99(1) European Patent Convention).
Printed by Jouve, 75001 PARIS (FR)
(19)
EP 0 888 345 B1
&
(11) EP 0 888 345 B1
(12) EUROPEAN PATENT SPECIFICATION
(45) Date of publication and mention
of the grant of the patent:
05.05.2010 Bulletin 2010/18
(21) Application number: 97917552.8
(22) Date of filing: 19.03.1997
(51) Int Cl.:
C07D 451/02
(2006.01)
C07D 401/00
(2006.01)
A61K 51/04
(2006.01)
(86) International application number:
PCT/US1997/004403
(87) International publication number:
WO 1997/034892 (25.09.1997 Gazette 1997/41)
(54) SIGMA-2 RECEPTORS AS BIOMARKERS OF TUMOR CELL PROLIFERATION
SIGMA-2 REZEPTOREN ALS BIOMARKIERER DER TUMORZELLENPROLIFERATION
RECEPTEURS SIGMA-2 UTILISES EN TANT QUE BIOMARQUEURS DE LA PROLIFERATION DES
CELLULES TUMORALES
(84) Designated Contracting States:
AT BE CH DE DK ES FI FR GB GR IE IT LI LU MC
NL PT SE
Designated Extension States:
AL LT LV RO SI
(30) Priority: 20.03.1996 US 13717 P
(43) Date of publication of application:
07.01.1999 Bulletin 1999/01
(73) Proprietor: Wake Forest University Health
Sciences
Winston-Salem, NC 27157-1023 (US)
(72) Inventors:
• MACH, Robert, H.
Winston-Salem, NC 27106 (US)
• WHEELER, Kenneth, T., Jr.
Winston-Salem, NC 27106 (US)
• YANG, Biao
Winston-Salem, NC 27103 (US)
• CHILDERS, Steven, R.
Winston-Salem, NC 27106 (US)
(74) Representative: Beresford, Keith Denis Lewis et al
Beresford & Co.
16 High Holborn
London
WC1V 6BX (GB)
(56) References cited:
WO-A-93/08185 WO-A-95/21820
US-A- 4 797 387 US-A- 4 808 588
US-A- 5 106 843 US-A- 5 330 990
US-A- 5 436 251
• KLINE R.H. ET AL.: ’Synthesis of 3-
Carbamoylecgonine Methyl Ester Analogues as
Inhibitors of Cocaine Binding and Dopamine
Uptake’ J. MED. CHEM. vol. 34, 1991, pages 702
- 705, XP002947712
• TURCONI M. ET AL.: ’Synthesis of a New Class
of 2,3-Dihydro-2-oxo-1H-benzimidazole-1-carbox
ylic Acid Derivatives as Highly Potent 5-HT3
Receptor Antagonists’ J. MED. CHEM. vol. 33,
1990, pages 2102 - 2108, XP002947713
• BERMUDEZ J. ET AL.: ’5-Hydroxytryptamine (5-
HT3) Receptor Antagonists. 3. Ortho-Substituted
Phenylureas’ J. MED. CHEM. vol. 33, 1990, pages
1932 - 1935, XP002947714
Remarks:
The file contains technical information submitted after
the application was filed and not included in this
specification

EP 0 888 345 B1
2
5
10
15
20
25
30
35
40
45
50
55
Description
Background of the Invention
[0001] Breast cancer is characterized by a proliferative potential that can vary considerably from patient to patient.
The rate of cell proliferation has been shown in breast tumors to predict the response to radiation therapy and chemo-
therapy. Presently, measures of cell proliferation are obtained by histological or flow-cytometric analysis. Both methods
are limited by sampling procedures and only 60-70% of patient samples are suitable for flow cytometric analysis.
[0002] It was recently demonstrated that sigma-2 (σ2) receptors are expressed in high density in a number of human
and rodent breast cancer cell lines (Cancer Research, 55, 408 (1995)). However, their expression is heterogenous, and
their function is unknown.
[0003] A continuing need exists for noninvasive methods that can accurately assess the proliferative status of breast
cancer. Such methods could have a significant impact on determining an optimal therapy for treating breast cancer
patients.
[0004] United States Patent No. 4,808,588 discloses heterocyclic ureas and carbonates useful as pharmaceuticals,
being compounds of the formula illustrated below, or pharmaceutically acceptable salts thereof:
wherein:
Het is monocyclic heteroaryl having two adjacent carbon atoms, a and b, depicted in formula (I); R1 and R2 are
independently selected from hydrogen, halogen, CF3, C1-6 alkyl and C1-6 alkoxy;
R3 is hydroxy, C1-6 alkoxy, C3-7 alkenyl-methoxy, phenoxy or phenyl C1-4 alkoxy in which either phenyl moiety may
be substituted by one or two C1-6 alkyl, C1-6 alkoxy or halo; CO2R6 wherein R6 is hydrogen or C1-6 alkyl, CONR7R8
or SO2NR7R8 wherein R7 and R8 are independently hydrogen or C1-6 alkyl or together are C4-6 polymethylene,
NO2, (CH2)mOR9 wherein m is 1 or 2 and R9 is C1-6 alkyl or S(O)nR10 wherein n is 0, 1 or 2 and R10 is C1-6 alkyl;
L is NH or O;
Z is a group of formula (a), (b) or (c);

EP 0 888 345 B1
3
5
10
15
20
25
30
35
40
45
50
55
wherein n is 2 or 3; p is 1 or 2; q is 1 to 3; r is 1 to 3; and
R4 or R5 is C1-4 alkyl.
[0005] United States Patent No. 5,106,843 discloses heterocyclic compounds, particularly aryl ureas and carbamic
acid derivatives of the general formula given below, which are useful as 5-HT3 antagonists:
A-NHCW-Y-B
wherein A is a specified aromatic radical including
3,5-dichlorophenyl;
W is O or S;
Y is NH or O, and
B is a specified saturated azacyclic ring such as tropan-3-yl or quinuclidin-3-yl.
Summary of the invention
[0006] The present invention provides a noninvasive method to detect cancer cells or to assess the proliferative status
of cancer cells which express sigma-2 (σ2) receptors, such as cells of solid tumors. The method preferably comprises
(a) ; contacting in vitro a cell sample of the tumour with an amount of a detectably labeled compound of formula (I):
wherein R is C6F5CH2, or T-C6H4CH2 wherein T is halo, CH3S, CH3O, NH2 or H; A is NH, O or S, B is NH, O or S; G
is O or S; D is CH or N; E is CH or N; F is CH or N; Y and Z are individually H, halo, OH, (C1-C4)alkyl, (C1-C4)alkoxy,
(C1-C4)alkylC(O), (C1-C4)alkylS, NH2, SH, N(R)2 or together are OCH2O, wherein said alkyl can be straight, branched,
cycloalkyl or (cycloalkyl)alkyl; X is (CH2)2, (CH2)3 or CH=CH; or a pharmaceutically acceptable salt thereof, wherein the
label comprises a radionulide; and (b) determining the extent to which the compound of formula (I) binds to the cells of
said tumour, said extent providing a measure of the proliferative status of said tumour, which status correlates to the
extent of sigma-2 receptor expression by said cells. The method is based on the ability of the compound of formula (I)
to selectively bind to sigma-2 (σ2) receptors, versus σ1 receptors.
[0007] Groups Z and Y can occupy any ring position, i.e., any one of E, D or F can CY or CZ. Preferably, at least one
of Y or Z is not H. Preferably A is O and B is NH. Preferably, R is CH, benzyl or phenyl. Alkyl can be branched, unbranched,
cycloalkyl or (cycloalkyl)alkyl.
[0008] The label is a radionuclide, selected from a radioisotope of halogen (125I, 123I, 18F, 19F) or 11C. The compound

EP 0 888 345 B1
4
5
10
15
20
25
30
35
40
45
50
55
is preferably administered parenterally, i.e., by intravenous, i.p., intrathecal or enteral administration.
[0009] Novel compounds of formula (I), labeled and unlabeled, are also within the scope of the invention, as are
pharmaceutical compositions comprising one or more of said compounds. The unlabeled compound can be used as
intermediates to make labeled compounds or as sigma-2 specific ligands which can be used in competitive assays to
assay for the presence of σ2 receptors, as described below. The configuration at the 3-position can be
exo
- or
endo
-;
of which
endo
- is preferred.
Brief Description of the Figures
[0010]
FIG. 1 shows compounds of the invention.
FIG. 2 shows compounds of the invention.
FIG. 3 shows compounds of the invention.
Detailed Description of the Invention
[0011] Processes for preparing compounds of formula 1 are illustrated by the following procedures in which the
meanings of the generic radicles are as given above unless otherwise qualified.
[0012] Compounds of formula I wherein A is oxygen and B is nitrogen can generally be prepared by reacting an
isocyanate or formula II with an alcohol of formula III under standard conditions.
Solvents, bases, and reaction conditions suitable for such a reaction are well known to the art. For example, the reaction
may conveniently be carried out under conditions similar to those described in Example 1.
[0013] Compounds of formula I wherein A is nitrogen and B is nitrogen can generally be prepared by reacting an
isocyanate of formula II with an amine of formula IV under standard conditions.
Solvents, bases, and reaction conditions suitable for such a reaction are well known to the art.
[0014] It is noted that many of the starting materials employed in the synthetic methods described above are com-
mercially available, are reported in the scientific literature, or can be prepared using methods analogous to those described
in the literature.
[0015] In cases where compounds are sufficiently basic or acidic to form stable nontoxic acid or base salts, adminis-
tration of the compounds as salts may be appropriate. Examples of pharmaceutically acceptable salts are organic acid
addition salts formed with acids which form a physiological acceptable anion, for example, tosylate, methanesulfonate,

EP 0 888 345 B1
5
5
10
15
20
25
30
35
40
45
50
55
acetate, citrate, malonate, tartarate, succinate, benzoate, ascorbate, α-ketoglutarate, and α-glycerophosphate. Suitable
inorganic salts may also be formed, including hydrochloride, sulfate, nitrate, bicarbonate, and carbonate salts.
[0016] Pharmaceutically acceptable salts may be obtained using standard procedures well known in the art, for example
by reacting a sufficiently basic compound such as an amine with a suitable acid affording a physiologically acceptable
anion. Alkali metal (for example, sodium, potassium or lithium) or alkaline earth metal (for example calcium) salts of
carboxylic acids can also be made.
[0017] The compounds of formula I can be formulated as pharmaceutical compositions and administered to a mam-
malian host, such as a human patient in a variety of forms adapted to the chosen route of administration, i.e., orally or
parenterally, by intravenous, intramuscular, topical or subcutaneous routes.
[0018] Thus, the present compounds may be systemically administered, e.g., orally, in combination with a pharma-
ceutically acceptable vehicle such as an inert diluent or an assimilable edible carrier. They may be enclosed in hard or
soft shell gelatin capsules, may be compressed into tablets, or may be incorporated directly with the food of the patient’s
diet. For oral therapeutic administration, the compound may be combined with one or more excipients and used in the
form of ingestible tablets, buccal tablets, troches, capsules, elixirs, suspensions, syrups, wafers, and the like. Such
compositions and preparations should contain at least 0.1% of the compound. The percentage of the compositions and
preparations may, of course, be varied and may conveniently be between about 2 to about 60% of the weight of a given
unit dosage form. The amount of compound in such therapeutically useful compositions is such that an effective dosage
level will be obtained.
[0019] The tablets, troches, pills, capsules, and the like may also contain the following: binders such as gum tragacanth,
acacia, corn starch or gelatin; excipients such as dicalcium phosphate; a disintegrating agent such as corn starch, potato
starch, alginic acid and the like; a lubricant such as magnesium stearate; and a sweetening agent such as sucrose,
fructose, lactose or aspartame or a flavoring agent such as peppermint, oil of wintergreen, or cherry flavoring may be
added. When the unit dosage form is a capsule, it may contain, in addition to materials of the above type, a liquid carrier,
such as a vegetable oil or a polyethylene glycol. Various other materials may be present as coatings or to otherwise
modify the physical form of the solid unit dosage form. For instance, tablets, pills, or capsules may be coated with gelatin,
wax, shellac or sugar and the like. A syrup or elixir may contain the compound, sucrose or fructose as a sweetening
agent, methyl and propylparabens as preservatives, a dye and flavoring such as cherry or orange flavor. Of course, any
material used in preparing any unit dosage form should be pharmaceutically acceptable and substantially non-toxic in
the amounts employed. In addition, the compound may be incorporated into sustained-release preparations and devices.
[0020] The present compounds may also be administered intravenously or intraperitoneally by infusion or injection.
Solutions of a compound or its salts can be prepared in water, optionally mixed with a nontoxic surfactant. Dispersions
can also be prepared in glycerol, liquid polyethylene glycols, triacetin, and mixtures thereof and in oils. Under ordinary
conditions of storage and use, these preparations contain a preservative to prevent the growth of microorganisms.
[0021] The pharmaceutical dosage forms suitable for injection or infusion can include sterile aqueous solutions or
dispersions or sterile powders comprising a labeled or unlabeled compound of formula I adapted for the extemporaneous
preparation of sterile injectable or infusible solutions or dispersions, optionally encapsulated in liposomes. In all cases,
the ultimate dosage form must be sterile, fluid and stable under the conditions of manufacture and storage. The liquid
carrier or vehicle can be a solvent or liquid dispersion medium comprising, for example, water, ethanol, a polyol (for
example, glycerol, propylene glycol, liquid polyethylene glycols, and the like), vegetable oils, nontoxic glyceryl esters,
and suitable mixtures thereof. The proper fluidity can be maintained, for example, by the formation of liposomes, by the
maintenance of the required particle size in the case of dispersions or by the use of surfactants. The prevention of the
action of microorganisms can be brought about by various antibacterial and antifungal agents, for example, parabens,
chlorobutanol, phenol, sorbic acid, thimerosal, and the like. In many cases, it will be preferable to include isotonic agents,
for example, sugars, buffers or sodium chloride. Prolonged absorption of the injectable compositions can be brought
about by the use in the compositions of agents delaying absorption, for example, aluminum monostearate and gelatin.
[0022] Sterile injectable solutions are prepared by incorporating the compound in the required amount in the appropriate
solvent with various of the other ingredients enumerated above, as required, followed by filter sterilization. In the case
of sterile powders for the preparation of sterile injectable solutions, the preferred methods of preparation are vacuum
drying and the freeze drying techniques, which yield a powder of the labeled or unlabled compound of formula (I) plus
any additional desired ingredient present in the previously sterile-filtered solutions.
[0023] For topical administration, the present compounds may be applied in pure form, i.e., when they are liquids.
However, it will generally be desirable to administer them to the skin as compositions or formulations, in combination
with a dermatologically acceptable carrier, which may be a solid or a liquid.
[0024] Useful dosages of the compounds of formula I can be determined by comparing their
in vitro
activity, and
in
vivo
activity in animal models. Methods for the extrapolation of effective dosages in mice, and other animals, to humans
are known to the art; for example, see U.S. Pat. No. 4,938,949.
[0025] Generally, the concentration of the compound(s) of formula I in a liquid composition, such as a lotion, will be
from about 0.1-25 wt-%, preferably from about 0.5-10 wt-%. The concentration in a semi-solid or solid composition such
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
1
/
24
100%