introduction à l`enzymologie

BCM 1502 Introduction à l’Enzymologie Page 1/12
INTRODUCTION À L'ENZYMOLOGIE
Les enzymes sont des macromolécules spécialisées qui
- catalysent les réactions biologiques
- transforment différentes formes d'énergie.
Les enzymes diffèrent des catalyseurs chimiques par leurs propriétés:
1 - Leur taux de réaction est plus élevé. Les enzymes ont un pouvoir
catalytique de 106 à 1012 fois supérieur aux réactions non catalysées, et de plusieurs
ordres de grandeur supérieur aux réactions catalysées par des réactifs chimiques.
2 - Les réactions enzymatiques se font dans des conditions relativement
douces (pH neutre, basse température etc).
3 - Les réactions enzymatiques sont très spécifiques, à la fois pour le substrat
et pour le type de réaction. La réaction est efficace à 100% et ne génère normalement
pas de produits secondaires.
4 - Les réactions enzymatiques peuvent être modulées. La régulation de
l'activité enzymatique peut s'effectuer de façon allostérique, par des protéines
régulatrices, par une modification covalente de l'enzyme, par une activation
protéolytique, par la quantité d'enzyme produite, etc.
Nomenclature:
Les enzymes sont classifiés selon la réaction qu'ils catalysent. Ils sont
désignés par un nom commun (carboxypeptidase A), un nom systématique
(peptidyl-L-amino acide hydrolase) et un numéro de classification (EC « Enzyme
Commission » 3.4.17.1). Ce numéro, qui est spécifique pour chaque enzyme, est tiré
d'un tableau qui donne les 6 principales classes de réaction que les enzymes peuvent
effectuer, puis les sous-classes et les sous-sous-classes.
1 = oxydoréductase Réaction oxydation - réduction
2 = transférase Transfère des groupements fonctionnels
3 = hydrolase Réactions hydrolytiques
4 = lyase Réaction d’élimination pour former
des liaisons doubles
5 = isomérase Isomérisation
6 = ligase Formation des liaisons avec hydrolyse ATP
donc, la carboxypeptidase porte le numéro de classe 3 (hydrolase), la sous-
classe 4 (lien peptidique), la sous-sous-classe 17 (métallo-carboxypeptidase) et 1
(numéro de l'enzyme dans cette série).

BCM 1502 Introduction à l’Enzymologie Page 2/12
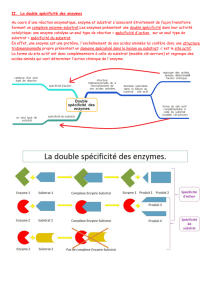
Spécificité pour le substrat.
Les interactions par lesquelles les substrats et autres molécules se lient à
l'enzyme sont les mêmes interactions qui dictent la conformation de la protéine elle-
même. Tous les deux cas impliqueront des interactions van der Waal,
électrostatiques, ponts hydrogènes, et hydrophobiques.
Le substrat se fixe au site actif de l'enzyme. Le site actif est défini à la fois
comme le site de fixation du substrat et le site catalytique. C'est donc un endroit
asymétrique, à la surface de la molécule, qui fixe le substrat et qui contient les acides
aminés nécessaires (ou groupes catalytiques) pour faire ou couper un lien. La
conformation et la composition chimique du site actif déterminent alors la spécificité
de l'enzyme – voir figure d’un complexe
enzyme-substrat montrant à la fois les
complémentarités géométrique et physique
entre enzyme et substrat. Les groupes
hydrophobiques sont symbolisés par un h dans
un cercle brun et les lignes en pointillés
figurent les liaisons hydrogènes.
On peut déterminer 5 caractéristiques du
site actif:
1 - Le site actif est une région restreinte
par rapport à la protéine totale
2 - Le site actif est une région
tridimensionnelle bâti à partir d'acides aminés
qui proviennent de différentes régions de la
chaîne polypeptidique.
3 - Le substrat se fixe au site actif par
des interactions faibles
4 - Les études cristallographiques des enzymes indiquent que le site actif est
préformé, c'est-à-dire qu'il existe à la surface des enzymes (hypothèse clé serrure),
même en absence du substrat. Cependant il est quelque peu flexible et s'ajuste après
fixation du substrat, selon l'hypothèse de l'induction de complémentarité ("induced
fit").
5 - La spécificité de l'association enzyme-substrat dépend de l'arrangement
précis des atomes au site actif.

BCM 1502 Introduction à l’Enzymologie Page 3/12
Co-facteur.
Plusieurs enzymes ont besoin de co-facteurs pour agir. Ces co-facteurs
peuvent être des ions (Fe2+, Mg2+, Zn2+, etc), des co-enzymes ou encore les deux à
la fois. Les co-enzymes sont des molécules organiques, souvent des vitamines, qui
servent de transporteurs de groupes fonctionnels ou d'électrons – voir structure de
NAD(P)+. Les ions, par contre, peuvent agir au site catalytique, servir de site de
liaison au substrat ou encore d'agents qui stabilisent la forme active de l'enzyme.
La terme l'holoenzyme décrit le complexe actif d'enzyme - co-facteur et
l'apoenzyme correspond à l'enzyme sans co-facteur et qui souvent ne possède pas
d'activité.
TAUX DE CATALYSE DES RÉACTIONS ENZYMATIQUES
Les enzymes sont des catalyseurs qui sont soumis aux lois de la
thermodynamique. Les lois de la thermodynamique nous permettent de déterminer
si une réaction peut se faire spontanément ou non. La cinétique enzymatique nous
permet de calculer le taux de catalyse, et comment ce taux peut être changé en
réponse à différentes conditions physiologiques et/ou pathologiques.
La cinétique enzymatique permet de découvrir
- l'affinité d'un substrat ou d'un inhibiteur pour un enzyme
- le taux de catalyse maximale
- l'efficacité enzymatique
- le mécanisme d'action des enzymes
- les variations de l'efficacité de l'enzyme dans différentes maladies
- la régulation des voies métaboliques
- le quantité d'enzyme présent dans un tissu
Cinétique chimique
La cinétique enzymatique est une sous discipline de la cinétique chimique et
par conséquence possède et utilise le même formalisme. Ce formalisme dépend des
notions suivantes:
Réactions élémentaires
Une réaction de stoechiométrie A P peut passer à travers une séquence de
réactions élémentaires de type A I1 I2 P. La caractérisation des réactions
élémentaires décrivant la réaction totale constitue la description mécanistique.

BCM 1502 Introduction à l’Enzymologie Page 4/12
Cofacteurs enzymatiques

BCM 1502 Introduction à l’Enzymologie Page 5/12
Les structures et les réactions du nicotinamide
adénine dinucléotide (NAD+) et du nicotinamide
adénine dinucléotide (NADP+). Leurs formes
réduites sont NADH et NADPH. Ces cofacteurs
sont des transporteurs intracellulaires d’équivalents
réducteurs (électrons). Seul le noyau nicotinamide
est modifié au cours de la réaction. La réduction
implique formellement le transfert de deux atomes
d’hydrogène (H).
Co-enzyme – NAD(P)+
(NADP+)
(NAD+)
 6
7
8
9
10
11
12
6
7
8
9
10
11
12
1
/
12
100%