Traitement médical de l`hypercorticisme

Frédéric Castinetti,
Bernard Conte-Devolx,
Thierry Brue
Service d’endocrinologie,
diabète et maladies métaboliques,
CHU La Timone,
Rue Saint Pierre,
13005 Marseille, France
Correspondance :
Thierry Brue
Service d’endocrinologie,
diabète et maladies métaboliques,
CHU La Timone,
Rue Saint Pierre,
13005, Marseille,
E-mail : Thierry[email protected]
Tél : 04 91 38 65 97
Fax : 04 91 38 45 42
Mots-clé :
Maladie de Cushing,
sécrétion ectopique d’ACTH,
corticosurrénalome,
adénomes hypophysaires,
ketoconazole,
mitotane,
mifepristone,
hypercortisolisme
L’hypercorticisme ou syndrome de
Cushing est une pathologie rare [1]. Les trai-
tements symptomatiques et surtout étiolo-
giques sont indispensables du fait du risque
de complications cardio-vasculaires, méta-
boliques et osseuses. Les modalités thérapeu-
tiques sont nombreuses, et varient en fonc-
tion de l’étiologie [2]. Ainsi, la chirurgie
transsphénoïdale est le traitement de réfé-
rence de la maladie de Cushing, mais n’est
efficace que dans 70 à 80 % des cas, impo-
sant une radiothérapie complémentaire ou
une surrénalectomie bilatérale. La chirurgie
reste également le traitement de première
intention des syndromes de Cushing malins,
comme par exemple les corticosurrénalomes
et les tumeurs endocrines avec sécrétion ecto-
pique d’ACTH. Cependant chimiothérapie
et/ou radiothérapie adjuvantes sont souvent
nécessaires mais peu efficaces [2]. Les traite-
ments anti-cortisoliques peuvent donc être
utiles dans les 4 situations suivantes :
- Pendant la phase de localisation d’une
tumeur endocrine sécrétant de l’ACTH.
- Pour diminuer l’hypercortisolisme avant
une chirurgie ou après une chirurgie ineffi-
cace.
- Après radiochirurgie ou radiothérapie en
attente de l’efficacité maximale de la procé-
dure.
- Dans le cadre de l’association de traitements
pour une pathologie métastatique.
De nombreux traitements anti-corti-
soliques sont utilisés dans le syndrome de
Cushing, chacun ayant des avantages et des
inconvénients [3]. Cette revue détaille les
résultats des principales études rapportant
l’efficacité et les effets secondaires des prin-
cipaux traitements anti-cortisoliques à effets
surrénalien ou central.
Traitement à effets
surrénaliens (Tableau)
La plupart de ces traitements (à l’exception
de l’etomidate) ont fait l’objet de nombreuses
publications, avec des résultats à court et
long-terme, permettant de définir leur place
dans la prise en charge des hypercorticismes.
Leur principal inconvénient (à quelques rares
exceptions) est qu’ils ont une action purement
suspensive avec une reprise plus ou moins
rapide de l’hypercorticisme à l’arrêt du traite-
ment. A long-terme, il est en général nécessaire
d’augmenter progressivement les doses de trai-
tement pour éviter un échappement : la dimi-
nution des concentrations de cortisol va en
effet entraîner une augmentation des concen-
trations d’ACTH qui va stimuler la sécrétion
tumorale. L’effet secondaire naturel est la surve-
nue d’une insuffisance surrénalienne, qui justi-
fie l’arrêt transitoire du traitement, l’instaura-
tion d’hydrocortisone, puis la reprise éventuelle
de l’anticortisolique à dose plus faible.
Mitotane (o,p’-DDD)
• Mode d’action
Le mitotane inhibe la stéroïdogenèse
via le cytochrome P450scc (side chain clea-
vage) porteur de l’activité 20,22 lyase, et la
11ß-hydroxylase. La drogue aurait aussi des
effets atrophiants surrénaliens retardés, expli-
quant qu’une partie des patients (10 à 30 %)
peut présenter une insuffisance surrénalienne
définitive après arrêt du traitement. Du fait
d’une action cytotoxique sur la cellule surré-
nalienne, le mitotane est le traitement de choix
des corticosurrénalomes, même si son utilisa-
tion a été étendue au traitement des hypercorti-
44 Médecine Clinique endocrinologie & diabète • Hors-Série •
SYNDROME DE CUSHING
Traitement médical
de l’hypercorticisme
Cusching Brue.indd 44 14/12/10 14:10

cismes ACTH-dépendants. L’AMM reste
limitée au traitement des corticosurré-
nalomes à un stade avancé.
• Posologie
Le mitotane se présente sous forme
de comprimés à 500 mg (Lysodren®).
La posologie varie de 0,5 à 8 g par jour
en fonction de l’efficacité et de la tolé-
rance. La zone entre efficacité et effets
secondaires est étroite, justifiant des
dosages répétés de la mitotanémie. Le
seuil d’efficacité est variable en fonction
des étiologies entre 10 et 15 mg/l.
• Efficacité
Plusieurs études ont rapporté une
efficacité anti-sécrétoire du mitotane
dans la majorité des cas de corticosur-
rénalomes traités. Ce contrôle peut
être suivi d’un échappement thérapeu-
tique en fonction du stade de la mala-
die. Terzolo et al. ont, en outre, rapporté
une augmentation de la survie chez les
patients traités par mitotane en tant que
traitement adjuvant après chirurgie, en
comparaison avec des patients sans trai-
tement adjuvant [4]. Dans les tumeurs
avec sécrétion ectopique d’ACTH, une
étude française a récemment rapporté
l’efficacité du mitotane : sur 23 patients
traités en moyenne pendant environ
2 ans (posologie, 3,3 ± 1,2 g/jour), une
normalisation des concentrations de
cortisol libre urinaire a été observée
dans 90 % des cas. La tumeur endocrine
initialement non visualisable a été iden-
tifiée dans 8 cas sur 13, suggérant que
le mitotane pourrait être utilisé pour
contrôler l’hypersécrétion, en atten-
dant d’identifier la tumeur causale [5].
Enfin, dans la maladie de Cushing,
plusieurs études ont rapporté une effi-
cacité anti-sécrétoire dans 60 à 80 %
des cas. L’efficacité anti-sécrétoire du
mitotane est donc globalement élevée,
et ce, quelle que soit l’étiologie de l’hy-
percorticisme [2]. A noter cependant
que l’efficacité est retardée, le plateau
thérapeutique étant en général obtenu
après plusieurs semaines de traitement.
A l’inverse, la drogue s’accumule dans le
tissu adipeux, et a des effets prolongés
même après arrêt du traitement (demi-
vie longue).
• Effets secondaires
Les effets sont variables, et en géné-
ral dose-dépendants : ils associent sur
le plan clinique, des troubles digestifs
(diarrhée, nausée) et des troubles neuro-
logiques (vertiges, confusion, paresthé-
sies) ; sur le plan biologique, l’utilisation
de mitotane entraîne une hypercho-
lestérolémie LDL, une hypertriglycéri-
démie, une cholestase, et des troubles
hématologiques (anémie, neutrocyto-
pénie). L’étude française portant sur les
patients traités pour sécrétion ectopique
d’ACTH a également souligné la grande
fréquence de survenue d’une insuffi-
sance surrénalienne (20 cas sur 23), et
la survenue d’effets secondaires dans
environ 50 % des cas [5].
• Surveillance
Le traitement est adapté en fonc-
tion des mitotanémies, et des concen-
trations de cortisol libre urinaire (les
concentrations de cortisol plasmatique
peuvent être biaisées par l’augmentation
des concentrations de CBG par le mito-
tane). Une contraception efficace doit
être donnée à toute femme en âge de
procréer pendant le traitement et dans
les 2 ans qui suivent son arrêt.
Un traitement par hydrocorti-
sone doit être associé pour éviter une
45
Médecine Clinique endocrinologie & diabète • Hors-Série •
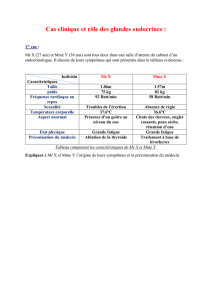
Tableau. Principaux traitements à visée anticortisolique.
Mécanisme Nom Posologie Efficacité
anti-sécrétoire
Effets
secondaires
Surrénalien Mitotane 500 à 8000 mg/j
(adaptation sur
mitotanémie,
CLU)
Elevée (70 à
80 %) quelle que soit
l’étiologie
Troubles digestifs
et neurologiques,
hypercholestéro-
lémie
Metyrapone 500 à 6000 mg/j
(adaptation sur
CLU)
Elevée (70-80 %)
quelle que soit
l’étiologie, mais
majorité des patients
rapportés traités
conjointement
par radiothérapie
Hyperandrogénie,
aggravation
de l’HTA et de
l’hypokaliémie
possibles
Ketocona-
zole
200 à 1600 mg/j
(adaptation sur
CLU)
Bonne (50 %) quelle
que soit l’étiologie
de syndrome de
Cushing
Risque
d’hépatite
fulminante
1/15000
Etomidate 2 à 4 mg/h IV
uniquement
(pas de voie orale
possible)
(adaptation sur
CLU)
Elevée quelle que
soit
l’étiologie
Surveillance
en réanimation;
insuffisance
surrénale
rapidement
observée
Central Octreotide
LP/
Lanreotide
SOM230
10 à 30 mg IM/
mois
60 à 120 mg/IM
ou s-cut profond/
mois
600 à 900 µg s-cut
2 fois/jour
Bonne (50 %) dans
les sécrétions
ectopiques d’ACTH
Etudes en cours sur
l’efficacité dans la
maladie de Cushing
Troubles digestifs,
lithiase biliaire
Cabergoline 1 à 7 mg/semaine Bonne (50 %) mais
études avec un faible
nombre de patients
traités
Troubles digestifs,
somnolence,
hypoTA,
valvulopathie ?
Antagonisme
des
récepteurs
aux glucocor-
ticoïdes
Mifepristone 200 à 1200 mg/j
(ACTH et cortisol
ininterprétables)
Elevée quelle que
soit l’étiologie
Risque
d’hypokaliémie
Cusching Brue.indd 45 14/12/10 14:10

insuffisance surrénalienne : la posolo-
gie requise est en général supérieure
d’un tiers à celle donnée pour les autres
causes d’insuffisance surrénalienne, car
le mitotane accélère le métabolisme
hépatique. A noter, cette accélération
du métabolisme hépatique modifie
également la demi-vie des traitements
anti-vitamine K.
• En pratique
Le mitotane est un traitement effi-
cace du syndrome de Cushing, quelle
que soit son étiologie. Il faut cepen-
dant souligner son efficacité retardée,
qui fera préférer d’autres traitements en
cas d’hypercorticisme massif sympto-
matique, et sa tolérance moyenne, qui
justifie une surveillance régulière des
mitotanémies.
Kétoconazole
• Mode d’action
Le kétoconazole (Nizoral®) est un
antifungique possédant des proprié-
tés anti-cortisoliques via l’inhibition
du cytochromes P450scc (side chain
cleavage) porteur de l’activité 20-22
lyase, de la 11ß-hydroxylase, et de la
17a-hydroxylase. Des effets extra-surré-
naliens ont également été décrits : effet
antagoniste du récepteur aux gluco-
corticoïdes en culture de cellules hépa-
tiques, liaison au récepteur aux gluco-
corticoïdes en cultures de monocytes.
En outre, une diminution des concen-
trations de CRH et d’ACTH a été obser-
vée en culture de cellules hypophy-
saires de syndrome de Nelson. Ces effets
extra-surrénaliens sont probablement
accessoires par rapport aux effets surré-
naliens. Le ketoconazole a également
des propriétés anti-androgéniques, ce
qui explique son utilisation en proto-
cole dans le traitement de cancers pros-
tatiques.
• Posologie
Le ketoconazole se présente sous
forme de comprimés à 200 mg. La dose
d’instauration du traitement varie de
200 à 600 mg/jour avec une augmenta-
tion progressive, adaptée à l’efficacité et
aux effets secondaires, jusqu’à 1600 mg/
jour répartis en 4 prises. Point impor-
tant à noter, l’utilisation du ketocona-
zole comme anticortisolique est hors
AMM. L’association aux statines est
contre-indiquée. Un cas clinique récent
a également rapporté une potentialisa-
tion des risques hémorragiques sous
warfarine.
• Efficacité
Plus de 100 patients traités par
ketoconazole ont été rapportés dans
la littérature [6, 7]. La majorité de ces
patients étaient porteurs d’une maladie
de Cushing. Quelques cas de patients
porteurs de corticosurrénalome ou de
sécrétion ectopique d’ACTH ont égale-
ment été rapportés sous forme de cas
cliniques ou de séries comportant un
faible nombre d’individus, avec une
efficacité anti-sécrétoire dans la majo-
rité des cas. La plupart de ces études
rapportent cependant des effets à court
terme du traitement.
Plus spécifiquement, dans la mala-
die de Cushing, une diminution des
concentrations de cortisol plasmatiques
a été rapportée dans 50 à 80 % des cas.
Dans notre série de 38 patients traités
en moyenne pendant 23 mois, plus de
la moitié étaient contrôlés par ketoco-
nazole à la dernière visite de contrôle ;
un quart présentaient une diminution
majeure des concentrations de cortisol
urinaires, sans être normalisés. De façon
surprenante, il n’existait pas de relation
entre la dose et l’efficacité : l’augmenta-
tion de posologie chez les patients en
échec après 3 mois de traitement ne
modifiait pas sensiblement le taux de
contrôle de l’hypersécrétion. Quinze
patients ont été traités car aucun
adénome hypophysaire n’était visible à
l’IRM: cinq d’entre eux ont finalement
bénéficié d’un geste chirurgical (après
12 à 30 mois de traitement) du fait de
la visualisation secondaire de l’adé-
nome (augmentation de volume liée
à la diminution des concentrations de
cortisol ?). Trois de nos patients ont été
traités pendant plus de 60 mois, avec un
bon contrôle de la cortisolémie et l’ab-
sence d’effets secondaires [8]. Enfin, une
étude portant sur 5 patients traités pour
une maladie de Cushing a rapporté une
diminution des concentrations d’ACTH
de 30 à 75 %, soulignant un effet égale-
ment central de la molécule [9].
• Effets secondaires
et surveillance
Le principal effet secondaire à
redouter, bien que rare, est l’hépatite
fulminante. Elle survient dans 1/15000
cas. Une augmentation modérée des
enzymes hépatiques est fréquente (5 à
10 % des cas) en début de traitement ou
lors de l’augmentation de posologie ;
elle ne nécessite pas d’adaptation du
traitement si l’augmentation reste limi-
tée à 2 à 3 fois la normale. Le ketocona-
zole peut également être à l’origine de
douleurs abdominales, de gynécomastie
ou de cycles irréguliers. La surveillance
se fait sur les concentrations de cortisol
plasmatique et le bilan hépatique. De
façon surprenante, les cas d’insuffisance
surrénalienne sous ketoconazole sont
très rares, ce qui est vraisemblablement
dû à un blocage partiel de la sécrétion
de cortisol et à l’augmentation progres-
sive de la cortisolémie liée à l’augmenta-
tion des concentrations d’ACTH. Cette
augmentation de la cortisolémie néces-
site souvent une augmentation progres-
sive des doses de ketoconazole en cas de
traitement prolongé.
• En pratique
Le ketoconazole est un anticortiso-
lique efficace qui peut être utilisé dans
toutes les étiologies de syndrome de
Cushing. La surveillance du bilan hépa-
tique est indispensable en début de trai-
tement, et lors des augmentations de
posologie. Le risque d’hépatite fulmi-
nante est faible, mais doit être craint en
cas de perturbation hépatique.
Metyrapone
• Mode d’action
La metyrapone (Métopirone®)
inhibe la 11ß-hydroxylase. Elle réduit
ainsi les concentrations de cortisol.
L’augmentation des concentrations
de 11 desoxycortisol et corticostérone
ne provoque pas de rétrocontrôle
majeur sur la sécrétion d’ACTH.
46 Médecine Clinique endocrinologie & diabète • Hors-Série •
Syndrome de Cushing
Cusching Brue.indd 46 14/12/10 14:10

L’augmentation d’ACTH peut être à
l’origine d’un pseudo-hyperaldosté-
ronisme et d’une hyperandrogénie. A
noter, l’AMM concerne uniquement
le traitement des hypercortisolismes
ACTH-indépendants (hors AMM pour
le traitement des hypercortisolismes
ACTH-dépendants).
• Posologie
La metyrapone se présente sous
la forme de comprimés dosés à
250 mg. Le traitement est en géné-
ral démarré à faible dose (750 mg/
jour) avec augmentation progres-
sive jusqu’à 6000 mg/jour. Il n’existe
pas d’interaction majeure, même si
la metyrapone pourrait potentialiser
les effets toxiques du paracétamol. La
surveillance et l’adaptation du traite-
ment s’effectuent sur les taux de corti-
sol libre urinaire.
• Efficacité
Le premier cas rapportant l’effica-
cité de la metyrapone dans le syndrome
de Cushing remonte à près de 50 ans.
Malgré cela, l’efficacité de la mety-
rapone à long-terme est difficile à
évaluer car la plupart des études sont
anciennes, avec des critères différents
de définition de contrôle de l’hypersé-
crétion, et surtout du fait d’un co-trai-
tement fréquent par radiothérapie
fractionnée [6]. La plus grande série
publiée à ce jour porte sur 91 patients,
la majorité ayant été traitée pour mala-
die de Cushing [10]. A court terme (1 à
16 semaines), la metyrapone permettait
un contrôle de l’hypersécrétion dans
75 % des cas avec une dose moyenne
de 2250 mg/j. A long-terme (27 mois
en moyenne), la metyrapone permet-
tait un contrôle de l’hypersécrétion
dans environ 80 % des cas, et ce, quelle
que soit l’étiologie. Il faut cependant
préciser que la majorité de ces patients
avaient été traités au préalable par
radiothérapie fractionnée. Même si
la période de traitement est courte en
comparaison de l’efficacité retardée de
la radiothérapie, on ne peut exclure
qu’une partie de la diminution des
concentrations soit liée à la radiothéra-
pie (et pas seulement à la metyrapone).
• Effets secondaires et surveillance
Le risque d’insuffisance surrénale,
et d’hirsutisme modéré est lié au méca-
nisme de bloc enzymatique induit
par le médicament. Le pseudo-hype-
raldostéronisme peut entraîner une
aggravation de l’hypertension arté-
rielle, et théoriquement une hypoka-
liémie, même si ces effets dose-dépen-
dants sont rarement rapportés dans la
littérature. A court terme, les patients
peuvent également présenter des
troubles digestifs mineurs, des cépha-
lées et des vertiges. Un risque d’alopé-
cie a été rapporté à long-terme [6].
• En pratique
La metyrapone n’est que peu utilisée
en France, au contraire par exemple de
la Grande-Bretagne. Elle a pourtant une
efficacité certaine dans la majorité des
cas, avec un délai d’action assez rapide
et une tolérance globalement bonne.
Etomidate
• Mode d’action
L’etomidate est un anesthésiant
dérivé imidazolé qui inhibe le cyto-
chrome P450Scc porteur de l’activité
20-22 lyase, et la 11ß-hydroxylase. Son
activité anti-cortisolique a été rappor-
tée dès le début des années 1980 sur des
sujets contrôles.
• Posologie
L’etomidate est administré à raison
de 2 à 4 mg/h IV, en soins intensifs,
sous stricte surveillance de la pression
artérielle et de la SaO2. Le principal
inconvénient de l’etomidate est donc
son mode d’administration, par voie
intraveineuse exclusive (voie veineuse
centrale).
• Efficacité
A ce jour, 19 patients (dont
2 enfants) traités par etomidate pour
un hypercorticisme ont été rappor-
tés dans la littérature, sous forme
de cas cliniques ou de petites séries
(6 patients). Les 17 patients adultes
étaient traités majoritairement après
inefficacité d’autres anti-cortisoliques
(metyrapone, ketoconazole), ou dans
l’objectif de contrôler rapidement l’hy-
percortisolémie avant une surrénalec-
tomie bilatérale. La diminution de la
cortisolémie était très rapide, survenant
au cours des 12-24 premières heures.
Récemment, Krakoff et al. ont traité un
patient porteur d’une sécrétion ecto-
pique d’ACTH (sans cible identifiable)
par une alternance ketoconazole/etomi-
date : l’etomidate a été instauré lors
d’un épisode de septicémie avec abcès
digestif et maintenu pendant la période
de soins intensifs (8 semaines), ce qui a
permis de normaliser la cortisolémie ;
le ketoconazole a été re-instauré après
sortie des soins intensifs [11]. A noter la
survenue d’une myoclonie transitoire
au cours du traitement chez ce patient.
• Effets secondaires et surveillance
L’efficacité majeure anti-cortiso-
lique de l’etomidate nécessite l’adjonc-
tion d’hydrocortisone pour éviter un
passage rapide en insuffisance surréna-
lienne. Une autre possibilité est d’ef-
fectuer une titration par des dosages
de cortisol plasmatique quotidiens et
d’adapter la posologie d’etomidate à ces
dosages [11,12]. L’effet anti-cortisolique
s’estompe rapidement après arrêt de la
molécule même si une étude a évoqué
un effet persistant [11].
• En pratique
L’etomidate doit être réservé aux
cas sévères d’hypercorticisme avec
inefficacité ou impossibilité d’utili-
sation d’autres anti-cortisoliques en
phase aiguë, ou chez les patients pour
lesquels il faut obtenir très rapidement
une normocortisolémie avant surréna-
lectomie bilatérale.
La mifepristone,
un antagoniste
des récepteurs aux
glucocorticoïdes
La mifepristone est un antagoniste
des récepteurs aux glucocorticoïdes.
Elle se lie également avec une forte affi-
47
Médecine Clinique endocrinologie & diabète • Hors-Série •
Cusching Brue.indd 47 14/12/10 14:10

nité aux récepteurs de la progestérone
et avec une affinité faible aux récep-
teurs des androgènes. Le mécanisme
d’action de la mifepristone est à l’ori-
gine d’une augmentation des concen-
trations d’ACTH et de cortisol plas-
matiques, ce qui rend impossible leur
utilisation dans la surveillance et l’adap-
tation du traitement [22].
Posologie
La mifepristone se présente sous
forme de comprimés à 200 mg. Le trai-
tement doit être débuté à faible posolo-
gie (200 à 400 mg/jour) avec augmenta-
tion progressive jusqu’à 800-1000 mg/
jour, selon l’efficacité clinique (HTA,
signes cliniques d’hypercorticisme) et
la survenue d’effets secondaires.
Efficacité
Trente-sept patients (présentant
diverses étiologies de syndrome de
Cushing) ont été rapportés dans la litté-
rature, le traitement ayant été instauré
en addition ou en substitution d’autres
anti-cortisoliques [23]. La durée du
traitement était en général inférieure
à 3 mois. Plus de 85 % des patients
ont présenté une diminution rapide
de leurs signes cliniques d’hypercor-
ticisme au cours du premier mois. La
moitié a présenté une amélioration de
leurs chiffres tensionnels et/ou de leurs
chiffres glycémiques (avec diminution
des doses d’insuline et/ou de la posolo-
gie du traitement antidiabétique oral).
Enfin, la majorité des rares patients
présentant une forme psychiatrique
ont été améliorés dès les premiers jours
de traitement [23].
Effets secondaires
et surveillance
Le risque d’hypokaliémie est majeur,
lié à une fixation du cortisol en excès
sur les récepteurs aux minéralocorti-
coïdes, ou à une stimulation directe de
la sécrétion d’aldostérone par l’ACTH
en excès. Plus d’un tiers des patients ont
présenté un épisode d’hypokaliémie,
ou l’aggravation d’une hypokaliémie
préexistante au cours de leur traitement
[23]. Ce même mécanisme physiopa-
thologique explique aussi la surve-
nue d’épisodes d’HTA chez certains
patients. Dans ce cas, la spironolac-
tone est en général efficace. Le second
risque est celui d’insuffisance surré-
nalienne, évalué à 16 % dans la litté-
rature. Une suspicion d’insuffisance
surrénalienne sous mifepristone pose
2 problèmes : l’impossibilité de confir-
mation biologique du diagnostic (corti-
sol plasmatique non interprétable), et
l’inefficacité du traitement par hydro-
cortisone. Le diagnostic est donc prin-
cipalement clinique, et le traitement
repose sur l’arrêt de la mifepristone,
et la prescription de dexamethasone
(1 mg pour 400 mg de mifepristone)
pendant au moins 48 heures [24]. Il
est possible de reprendre le traitement
par la suite, mais à doses plus faibles,
sous surveillance. La surveillance se fera
donc essentiellement sur la recherche
de signes cliniques d’insuffisance surré-
nalienne, la mesure de la TA et le dosage
de la kaliémie. Enfin, du fait de son
mécanisme anti-progestatif, la mife-
pristone peut être à l’origine d’une
hyperplasie endométriale avec métror-
ragies, à surveiller par une échographie
pelvienne annuelle en cas de traitement
prolongé.
En pratique
Du fait du faible nombre de données
publiées, en particulier sur l’efficacité à
long terme, la mifepristone ne peut pas
être recommandée en 1ere intention de
traitement anti-cortisolique. Son utili-
sation devrait être réservée à l’ineffica-
cité ou la contre-indication des autres
anti-cortisoliques, ou, par exemple,
en association dans la prise en charge
d’un corticosurrénalome malin métas-
tatique. A noter, la rapidité d’efficacité
qui pourrait être un atout majeur dans
la prise en charge des formes psychia-
triques de syndrome de Cushing.
Traitements
à effets centraux
(sécrétion d’ACTH)
Les données sur les traitements
ayant une action centrale sont limitées
en comparaison avec les études publiées
sur les drogues à effets surrénaliens. La
plupart sont des cas cliniques ou des
études avec un effectif limité et/ou un
suivi relativement court.
Agonistes dopaminergiques
• Cabergoline
La cabergoline est un agoniste
dopaminergique qui constitue le trai-
tement de référence des prolactinomes.
Quelques études récentes ont rapporté
l’efficacité de la cabergoline dans la
maladie de Cushing [13]. Pivonello
et al. ont traité 20 patients porteurs
d’une maladie de Cushing pour inef-
ficacité d’une chirurgie transsphénoï-
dale [14]. La cabergoline était donnée à
raison de 1 mg/semaine avec augmen-
tation progressive à raison de 1 mg/
mois jusqu’à 7 mg/semaine ou jusqu’à
normalisation des concentrations de
cortisol libre urinaire. A 3 mois, le corti-
sol libre urinaire était normalisé chez
10 patients. A 18-24 mois, 8 patients
(avec une dose moyenne de 3,5 mg/
semaine) conservaient une valeur
normale de cortisol libre urinaire sous
cabergoline. Deux patients ont présenté
un échappement thérapeutique à
12 et 18 mois de traitement. La caber-
goline était généralement bien tolérée :
2 patients ont du arrêter leur traitement
pour hypotension artérielle sévère [14].
D’autres études (avec un faible effec-
tif) ont également rapporté une effi-
cacité dans environ 50 % des cas de
patients traités par cabergoline, ce qui
souligne le rôle potentiel de cette molé-
cule dans la maladie de Cushing. Une
étude a rapporté l’efficacité d’un traite-
ment conjoint cabergoline (2 à 3 mg/
semaine) – ketoconazole (200 à 400 mg/
jour) : il est cependant difficile de déter-
miner le rôle précis d’une molécule par
rapport à l’autre dans le contrôle de
l’hypercortisolisme.
Plusieurs études portant sur le trai-
tement dopaminergique de la maladie
de Parkinson ont rapporté un risque de
valvulopathie [15]. Les données sont
contradictoires sur un tel risque chez
les patients traités pour un prolacti-
nome, mais ce risque semble faible : les
doses sont très inférieures à celles utili-
sées pour la maladie de Parkinson. Une
surveillance échographique annuelle
48 Médecine Clinique endocrinologie & diabète • Hors-Série •
Syndrome de Cushing
Cusching Brue.indd 48 14/12/10 14:10
 6
6
1
/
6
100%