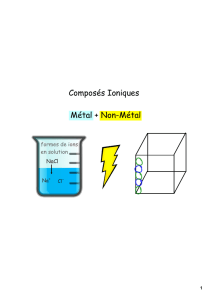
Application des liquides ioniques à la valorisation des métaux

Application des liquides ioniques `a la valorisation des
m´etaux pr´ecieux par une voie de chimie verte
Emmanuel Billy
To cite this version:
Emmanuel Billy. Application des liquides ioniques `a la valorisation des m´etaux pr´ecieux
par une voie de chimie verte. Autre. Universit´e Grenoble Alpes, 2012. Fran¸cais. <NNT :
2012GRENI009>.<tel-00721825>
HAL Id: tel-00721825
https://tel.archives-ouvertes.fr/tel-00721825
Submitted on 30 Jul 2012
HAL is a multi-disciplinary open access
archive for the deposit and dissemination of sci-
entific research documents, whether they are pub-
lished or not. The documents may come from
teaching and research institutions in France or
abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est
destin´ee au d´epˆot et `a la diffusion de documents
scientifiques de niveau recherche, publi´es ou non,
´emanant des ´etablissements d’enseignement et de
recherche fran¸cais ou ´etrangers, des laboratoires
publics ou priv´es.

THÈSE
Pour obtenir le grade de
DOCTEUR DE L’UNIVERSITÉ DE GRENOBLE
Spécialité : 2MGE Matériaux, Mécanique, Génie Civil,
Electrochimie
Arrêté ministériel : 7 août 2006
Présentée par
Emmanuel BILLY
Thèse dirigée par Farouk TEDJAR et
codirigée par Eric CHAÎNET
préparée au sein du Laboratoire du LEPMI de l’Université de
Grenoble et de l’entreprise RECUPYL à Domène
dans I-MEP2 (Ingénierie- Matériaux Mécanique Energétique
Environnement Procédés Production)
Application des liquides ioniques
à la valorisation des métaux
précieux par une voie de chimie
verte
Thèse soutenue publiquement le
10 Février 2012, devant le jury
composé de :
Pr Jean-Yves SANCHEZ
Professeur, Responsable d’équipe LEPMI, Saint Martin d’Hères, Président
Pr Jean-Yves HIHN
Professeur, Responsable d’équipe à l’Institut UTINAM, Besançon, Rapporteur
Dr Elisabeth CHASSAING
Directeur de recherche, IRDEP ParisTech, Rapporteur
Ing Marie APRIL
Ingénieur filières de recyclage à l’ADEME, Angers, Membre invité
Pr Farouk TEDJAR
Président de RECUPYL, Domène, Directeur de thèse
Dr Eric CHAÎNET
Chargé de recherche, Responsable d’équipe LEPMI, Saint Martin d’Hères,
Directeur de thèse


Remerciements
Je voudrai tout d’abord remercier chaleureusement mes directeurs de thèse, Farouk Tedjar et
Eric Chaînet de m’avoir fait partager leurs expériences, de la liberté d’action et d’autonomie
dont j’ai bénéficié durant ces trois années de thèse. J’aimerai également associer les personnes
de l’ADEME qui nous ont fait confiance et sans qui il m’aurait été impossible de faire cette
thèse. Par ailleurs, j’aimerai remercier mon jury de thèse Monsieur Jean-Yves SANCHEZ,
Monsieur Jean-Yves HIHN, Madame Elisabeth CHASSAING et Madame Marie APRIL pour
avoir accepté de juger de la qualité de ce travail.
Un merci tout particulier à Guy Espagnac, pour son aide et son enseignement dans l’art de la
verrerie. Je tiens également à remercier Denise Foscallo, Laure Cointreaux, Jean-Claude
Leprêtre et plus largement l’équipe ELSA et leurs doctorants pour leur aide et leur sympathie
durant ces trois années de thèse. Le bon déroulement de cette thèse n’est pas étranger à leur
soutien.
Je tiens à remercier toutes les personnes de RECUPYL que j’ai pu côtoyer au cours de ma
thèse. Un grand merci à Cristelle Pasquier qui à l’approche de la fin de ma thèse a été d’une
aide précieuse.
J’aimerai remercier toutes les personnes administratives du laboratoire qui nous
accompagnent et nous mettent dans les meilleurs dispositions pour atteindre nos objectifs :
Augustine Alessio pour sa patience et sa gentillesse et qui sans nul doute est un piler de
l’école doctorale, sans oublier Jacques Fouletier, qui de part ces qualités humaines et sa
proximité naturelle avec les étudiants a contribué au bon déroulement de ma thèse.
J’exprime tous mes remerciements aux personnes du CMTC qui sont bien souvent dans
l’ombre, mais qui réalisent toujours un travail fabuleux.
Merci à toute l’équipe ESME : Fredéric, Marian, Eric, Yvonne, Pascal, Laetitia, Benoît,
Julien, Zuzhen, Maguy, les anciens, Thiago, Bruno, … plus largement au Brésil et à notre
fantasque directeur de laboratoire Ricardo qui nous a fait partager avec ses stagiaires et
thésards la bonne humeur brésilienne.

Un grand merci à vous, ce fut un plaisir de vous faire le café tout les matins ;) J’ai fait la
connaissance de collègues, de brillants scientifiques mais surtout d’amis. Enfin, il y a une
personne que je souhaite tout particulièrement remercier, c’est l’ISE prize 2011, Monsieur
Frédéric Maillard qui depuis mon DRT m’a beaucoup apporté et m’a toujours conseillé de la
meilleure des façons.
J’aimerai remercier, comme j’aime à l’appeler ma troisième directrice de thèse,
Gipsy Lopez Billy. Elle m’a soutenue, aidée, écoutée et corrigée. Elle a été et est pour moi, ce
que l’atome est à l’électron, un lien indispensable pour son bon équilibre.
Enfin, je remercie toutes celles et ceux qui ont contribué de près ou de loin au bon
déroulement de mon travail ; je pense notamment à Muriel, Karine, Lenka, Laurent
Davoust,... ainsi qu’à tous les membres du LEPMI. Encore merci à vous qui avez, par votre
sympathie et votre bonne humeur, rendu ce travail si agréable et si enrichissant.
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
192
193
194
195
196
197
198
199
200
201
202
203
204
205
206
207
208
209
210
211
212
213
214
215
216
217
218
219
220
221
222
223
224
225
226
227
228
229
230
231
232
233
234
235
236
237
238
239
240
241
242
243
244
245
246
247
248
249
250
251
252
253
254
255
256
257
258
259
260
261
262
263
264
265
266
267
268
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
192
193
194
195
196
197
198
199
200
201
202
203
204
205
206
207
208
209
210
211
212
213
214
215
216
217
218
219
220
221
222
223
224
225
226
227
228
229
230
231
232
233
234
235
236
237
238
239
240
241
242
243
244
245
246
247
248
249
250
251
252
253
254
255
256
257
258
259
260
261
262
263
264
265
266
267
268
1
/
268
100%