Troubles du métabolisme lipidique de l`enfant

20
Vol. 24 No. 4 2013
Formation continue
Résumé
Les troubles du métabolisme lipidique sont
un groupe hétérogène de maladies généra-
lement héréditaires, diagnostiquées souvent
déjà chez l’enfant en raison d’une anamnèse
familiale positive. Il s’agit d’une tâche pédia-
trique de prévention secondaire importante;
le diagnostic et le traitement précoces de
certaines dyslipidémies conditionnent en
effet de façon décisive la santé à l’âge
adulte. Dans cet article nous souhaitons
aborder surtout les aspects pratiques, à
partir de questions souvent posées sur les
dyslipidémies, dont nous décrivons en bref
les plus fréquentes, ainsi que leur diagnostic
et traitement. Certains troubles du métabo-
lisme plus rares sont spécifiquement men-
tionnés, en renonçant néanmoins à les
traiter tous systématiquement. Les auteurs
de cet article estiment important pour la
pratique de ne pas se concentrer unique-
ment sur l’abaissement du taux de cholesté-
rol plasmatique mais de prendre en considé-
ration tous les facteurs de risque connus,
comme l’alimentation, l’activité physique, le
poids et le tabagisme. Pour le diagnostic
primaire autant que pour l’appréciation de la
situation globale la connaissance exacte de
l’anamnèse familiale est d’une importance
primordiale. Le traitement des dyslipidémies
est complexe et inclut la stimulation de
l’activité physique et l’évitement de facteurs
de risque tout autant que le régime et les
médicaments.
Introduction
Les troubles du métabolisme lipidique sont
parmi les maladies héréditaires les plus fré-
quemment rencontrées en pédiatrie et sont
d’une grande importance pour la santé à l’âge
adulte. En même temps ils ne provoquent
généralement pas de symptômes chez l’en-
fant, un fait qui ne doit en aucun cas inciter à
l’abstinence diagnostique et thérapeutique.
Bien au contraire, la pédiatrie joue un rôle
important en aiguillant sur une prise en
charge à vie adaptée et, si nécessaire, stricte
des troubles du métabolisme lipidique.
Vu sous cet angle, ce sujet est un parfait
exemple d’une des tâches les plus impor-
tantes en pédiatrie, la prévention secondaire.
Dans cette contribution l’accent est mis sur
les aspects pratiques, avec l’objectif d’en faci-
liter l’approche par le pédiatre praticien. Nous
répondons aux questions suivantes, posées
par un collègue praticien:
• Quand le pédiatre (praticien) doit-il envisa-
ger de contrôler le taux sanguin des lipides?
• Comment prendre en compte l’âge, la cli-
nique, l’anamnèse familiale et les facteurs
de risque?
• Quels paramètres contrôler et comment les
apprécier?
• Existent-ils des valeurs de référence et
comment les apprécier?
• Comment procéder lorsqu’on constate des
valeurs pathologiques? Quand les contrôler
et quand introduire un régime ou un traite-
ment médicamenteux?
L’article se concentre sur les situations fré-
quentes; les maladies plus rares ne sont pas
énumérées systématiquement et de manière
exhaustive, mais servent à illustrer certaines
situations cliniques ou biochimiques.
Définition
Les troubles du métabolisme lipidique (syno-
nyme: dyslipidémie) sont un groupe hétéro-
gène de maladies caractérisées par une ano-
malie des taux des lipides plasmatiques,
surtout du cholestérol et/ou des triglycérides.
Il peut s’agir d’une anomalie primaire en pré-
sence d’une dyslipidémie héréditaire ou se-
condaire dans le cadre d’une autre maladie
affectant le métabolisme hormonal (p.ex.
diabète, hypothyroïdie, syndrome de Cu-
shing), le rein (syndrome néphrotique ou une
autre maladie chronique du rein) ou le foie
(stéatose hépatique) ou d’une anorexie men-
tale. La plupart des dyslipidémies repré-
sentent un facteur de risque significatif pour
l’apparition d’une artériosclérose et doivent
donc être prises aux sérieux aussi chez l’en-
fant, bien que pour ainsi dire toujours asymp-
tomatique1)–3).
Épidémiologie
La dyslipidémie la plus fréquente est due à un
défaut hétérozygote du récepteur LDL4),
dont l’incidence est estimée à 1:500. Dans le
même ordre de grandeur se situe l’incidence
du déficit en apolipoprotéine B 100 (1:200–
1:700)qui engendre une affinité diminuée au
récepteur LDL5). Le défaut homozygote du
récepteur LDL, avec pour conséquence une
défaillance de facto de la fonction du récep-
teur LDL est par contre très rare (de l’ordre
de 1:1’000’000). Il faut préciser qu’il n’existe
pas de chiffres fiables concernant ces mala-
dies pour la Suisse. C’est le cas aussi pour
tous les autres troubles du métabolisme lipi-
dique, qu’il faut considérer rares ou très rares.
Cela signifie que pour la plupart de ces mala-
dies (exception faite pour les deux premières
mentionnées) on ne trouve en Suisse que très
peu de patients, parfois un seul voire aucun.
Biochimie et physiopathologie
En raison de leurs propriétés hydrophobes,
les lipides ne peuvent être transportés dans
le sang que s’ils sont fixés à une protéine
(apolipoprotéine). Dans la lipoprotéine qui en
résulte les lipides hydrophobes sont «cachés»
dans le noyau et entourés d’apolipoprotéines.
D’après leur densité on divise les lipopro-
téines en 5 classes, dont la composition ca-
ractéristique conditionne les propriétés bio-
chimiques6):
• Lipoprotéines high-density (HDL)
• Lipoprotéines low-density (LDL)
• Lipoprotéines intermediate-density (IDL)
• Lipoprotéines very low-density (VLDL)
• Chylomicrons
Ont une importance clinique surtout les LDL,
responsables du transport des lipides, surtout
du cholestérol, dans les organes périphé-
riques7). Une augmentation du taux plasma-
tique de LDL résulte d’un défaut du récepteur
LDL, comme c’est le cas dans l’hypercholes-
térolémie familiale. Outre le cholestérol, les
Troubles du métabolisme lipidique
de l’enfant
Johannes Häberle1), Alexander Lämmle1), Matthias R. Baumgartner1)
Traduction: Rudolf Schlaepfer, La Chaux-de-Fonds
1) Abteilung für Stoffwechselkrankheiten
Forschungszentrum für das Kind
Universitäts-Kinderspital Zürich
Steinwiesstrasse 75
CH-8032 Zürich

21
Vol. 24 No. 4 2013 Formation continue
LDL contiennent en abondance de l’apolipo-
protéine B 100, dont la fonction peut être
perturbée par des mutations génétiques
(p.R3500Q étant la mutation la plus fré-
quente)3 ) , 5) . Du point de vue clinique et théra-
peutique la situation (hypercholestérolémie)
qui en résulte est identique à celle d’un défaut
du récepteur LDL. Des taux élevés surtout de
cholestérol LDL peuvent engendrer des pa-
thologies vasculaires sous forme d’athéros-
clérose déjà pendant l’enfance.
Les défauts de la lipoprotéine lipase, qui
hydrolyse dans l’endothélium capillaire les
triglycérides contenus dans les chylomicrons
et les VLDL et permet ainsi leur absorption
dans les cellules, mènent à une augmentation
parfois massive des triglycérides (taux parfois
> 50 mmol/l, norme < 2 mmol/l). Les défauts
du cofacteur apolipoprotéine CII présentent
un phénotype identique. Les taux élevés de
triglycérides ne représentent pas un risque
d’athérosclérose précoce.
Lors de défauts du transporteur ABC G5 ou
G8, l’élimination dans l’intestin grêle et les
voies biliaires de graisses végétales est per-
turbée, les phytostérols étant résorbés aupa-
ravant. Il en résulte la très rare sitostéro-
lémie qui représente, comme le défaut homo-
zygote du récepteur LDL, un risque significatif
d’infarctus cardiaques déjà pendant l’en-
fance8).
Tableau clinique
La plupart des patients avec une dyslipidémie
sont asymptomatiques durant l’enfance. Des
dépôts graisseux dans la peau, les xanthé-
lasmes, peuvent être les premiers symp-
tômes de l’hypercholestérolémie familiale.
Dans le cas d’un défaut homozygote du récep-
teur LDL ils peuvent être visibles, déjà pen-
dant la petite enfance, sous forme de nodules
impressionnants le long des plis cutanés des
mains et des pieds et sur les articulations,
alors que les premiers dépôts jaunâtres se
trouvent généralement dans la paupière
inférieure4), 9) (Fig. 1A). L’expérience clinique
montre qu’ils corrèlent clairement avec le
taux plasmatique du cholestérol et qu’ils ré-
gressent, voire disparaissent, sous traite-
ment.
Certaines dyslipidémies (p.ex. sitostérolémie)
se manifestent par des dépôts graisseux
sous-cutanés plus profonds, les xanthomes.
Les localisations de prédilection sont les
faces d’extension du coude et du genou ainsi
que le tendon d’Achille (Fig. 1B). Souvent la
peau au-dessus des xanthomes présente une
coloration bleuâtre, livide qui persiste après
régression des xanthomes sous traitement.
L’arc sénile (arcus corneae) ne s’observe
qu’exceptionnellement pendant l’enfance, en
cas d’hypercholestérolémie mal compensée
(ou hypercholestérolémie homozygote).
Chez un petit nombre de patients l’accumula-
tion de graisse provoque une hépatomégalie
et, par distension de la capsule de Glisson,
des douleurs abdominales. Cette situation
se rencontre p.ex. chez les patients avec une
hypertriglycidémie familiale ou une chylomi-
cronémie familiale.
Le syndrome métabolique, actuellement en-
core relativement rare chez l’enfant est carac-
térisé par une obésité, une hyperuricémie,
une hypertension artérielle, une résistance
périphérique à l’insuline et un taux abaissé de
cholestérol HDL.
Complications
Les taux plasmatiques élevés de cholestérol
n’occasionnent pas de complications aiguës,
mais sont un facteur de risque pour une athé-
rosclérose précoce. Le poids de l’hypercho-
lestérolémie dans le contexte d’autres fac-
teurs de risque connus (surpoids, manque
d’activité physique, hypertension artérielle,
hyperhomocystinémie, taux élevé de lipopro-
téine [a], tabagisme) et inconnus est loin
d’être clair1). Des cas, rencontrés aussi dans
la pratique, d’hypercholestérolémie relative-
ment discrète à évolution fatale précoce
comme des familles à peine touchées par une
hypercholestérolémie pourtant sévère sont
décrits dans la littérature. On craint particu-
lièrement les complications en cas d’hyper-
cholestérolémie familiale homozygote et de
sitostérolémie, maladies pour lesquelles ont
été décrits des infarctus cardiaques aigus à
l’issue fatale déjà pendant l’enfance3), 8).
Des taux très élevés de triglycérides (>10
mmol/l) peuvent déclencher des symptômes
aigus: douleurs abdominales aiguës, hémor-
ragies gastro-intestinales et une pancréatite
aiguë dont le pronostic peut être sévère.
L’hypertriglycéridémie n’est par contre pas un
facteur de risque pour une athérosclérose
précoce.
Transition et perspectives
pour l’adulte
Les troubles du métabolisme lipidique restent
un domaine de la médecine adulte, pratique-
ment toutes les complications n’apparaissant
qu’à l’âge adulte. Idéalement les pédiatres et
médecins d’adultes maintiennent des
contacts étroits afin de garantir une transition
optimale et éviter ainsi des angoisses chez les
patients et les pertes d’informations par les
médecins. Mais, comme nous l’avons expli-
cité, la prise en charge pédiatrique compé-
tente et conséquente permet pour le moins
de retarder l’apparition de complications.
Anamnèse familiale
Sans doute une anamnèse familiale minu-
tieuse et complète joue un rôle primordial
lorsqu’on évalue la situation d’un patient avec
une dyslipidémie. Tous les parents au premier
degré sont au moins à inclure. Une anamnèse
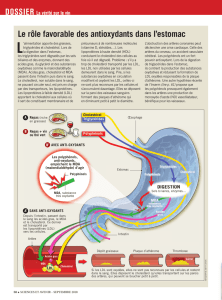
Figure 1: altérations cutanées lors de troubles du métabolisme lipidique. A: patiente de 9 ans avec des xanthélasmes que l’on reconnaît par la
présence de colorations jaunâtres sous les deux paupières inférieures. B: patient de 12 ans avec un xanthome sur le genou, apparaissant comme
une tuméfaction visible et palpable avec couleur bleuâtre de la pau.
A B

22
Vol. 24 No. 4 2013
Formation continue
ciblée avec des questions fermées est indis-
pensable («est-ce que vos parents ont subi un
infarctus précoce?», «… et leurs frères et
sœurs?» etc), des données importantes pou-
vant sinon se perdre. Ainsi infarctus, attaque
cardiaque, pontage p.ex. devraient être des
mot-clé10). Lorsque l’anamnèse familiale
s’avère positive, une analyse ciblée du profil
des lipides plasmatiques est indiquée déjà
chez l’enfant.
Examens de laboratoire
Lors de l’évaluation initiale d’une dyslipidémie
on dose les paramètres plasmatiques suivants
chez le patient à jeun3):
• Profil lipidique (cholestérol total, HDL, LDL
et triglycérides)
• Lipoprotéine (a) (la valeur est déterminée
génétiquement et ne nécessite donc pas de
contrôles, sauf lors d’un essai de traitement
avec la niacine)11)
• Homocystéine
Pour apprécier les taux des lipides il faut des
valeurs de référence en fonction de l’âge,
puisque notamment le taux de cholestérol
augmente pendant l’enfance et après la pu-
berté, alors que les valeurs peuvent sponta-
nément diminuer avant et pendant la puber-
té3).
Dans des situations particulières il peut s’avé-
rer raisonnable de procéder à des examens
spéciaux, telle l’électrophorèse des lipides ou
le profil des stérols; dans ces situations il est
conseillé de contacter un centre du métabo-
lisme.
Afin d’exclure un trouble secondaire du méta-
bolisme lipidique on dose, une seule fois, les
paramètre suivants:
• TSH, fT4
• Créatinine
• Cortisol
• Status urinaire
Traitement –
considérations générales
En principe presque tous les enfants avec une
dyslipidémie ont besoin d’un régime et/ou
d’un traitement médicamenteux. Il est par
contre difficile de décider à partir de quelle
modification des valeurs de laboratoire un
régime seul ne suffit plus et la prescription de
médicaments devient nécessaire. Cela ne peut
se décider qu’en tenant compte de la situation
globale, de l’âge, de la clinique (est-ce qu’il y
a d’autres facteurs de risque?) et de l’anam-
nèse familiale et ne se base jamais simple-
ment sur des paramètres biochimiques. En
principe la simple augmentation de l’activité
physique, à moins qu’elle ne soit déjà adé-
quate, peut améliorer le profil lipidique3), 4),
12 ) –14 ) .
Régime
La modification des habitudes alimentaires
est la pierre angulaire du traitement de la
plupart des dyslipidémies de l’enfant. Pour
l’hypercholestérolémie familiale cela signifie
une diminution des graisses animales satu-
rées et l’évitement des aliments particulière-
ment riches en cholestérol (surtout le jaune
d’œuf, la viande avec graisse visible, le
beurre). L’apport en lipides consistera de
préférence en graisses mono-insaturées
(p.ex. huile de noix, de colza ou d’olive) tout
en limitant leur proportion à 30–35% de l’ap-
port total en calories. En présence d’une si-
tostérolémie par contre, on évitera les ali-
ments riches en graisses végétales, alors que
les graisses animales sont «autorisées». Pour
certaines maladies, p.ex. l’hypertriglycidémie
familiale, on obtient un effet thérapeutique
suffisant en réduisant la proportion des
graisses dans l’alimentation à < 25% et en
évitant une consommation excessive de
sucres. Le résultat du régime n’est par contre
souvent pas satisfaisant pour l’hypercholes-
térolémie familiale; même appliqué rigoureu-
sement, il ne permet qu’une réduction de 10 %
jusqu’à un maximum de 20% du taux plasma-
tique du cholestérol, ce qui est insuffisant
pour de nombreux patients3).
L’efficacité d’un régime pauvre en cholestérol
est en partie déterminée génétiquement. Ce
sont notamment les phénotypes de l’apolipo-
protéine E qui servent de prédicteurs d’une
bonne ou mauvaise réponse au régime15).
Médicaments
Plusieurs médicaments sont disponibles, aux
mécanismes d’action différents. Pour plu-
sieurs raisons le choix se réduit, dans la pra-
tique, à un nombre restreint de substances
actives. Le traitement médicamenteux se fait
en principe toujours en poursuivant le ré-
gime3).
Les résines échangeuses d’ions (p.ex. co-
lestyramine) sont de grandes molécules inso-
lubles dans l’eau, non résorbées dans l’intes-
tin et possédant une grande affinité pour les
acides biliaires, réduisant ainsi la résorption
des lipides. Pratiquement cela ne marche que
si le médicament est pris lors de chaque re-
pas contenant des graisses. Ce fait et la
sensation désagréable en l’ingérant ex-
pliquent la mauvaise compliance (compréhen-
sible), réduisant ultérieurement l’efficacité
déjà limitée des résines échangeuses d’ions.
Bien que des recommandations de consensus
prévoient encore l’utilisation des résines
échangeuses d’ions comme traitement médi-
camenteux de première intention, dans leur
pratique les auteurs de cet article ne les
considèrent comme option acceptable que
pour de rares patients.
Les inhibiteurs de la HMG-CoA réductase
(synonyme: statines) influencent la synthèse
endogène du cholestérol à un stade très pré-
coce et réduisent de ce fait la disponibilité
intracellulaire du cholestérol. En même temps
la sensibilité des récepteurs LDL est renfor-
cée, ce qui augmente l’absorption de choles-
térol-LDL par la cellule. Les statines repré-
sentent donc un moyen efficace pour réduire
le taux de cholestérol total et LDL, la diminu-
tion pouvant atteindre, selon le médicament,
jusqu’à 40%. Dans tous les cas la combinai-
son avec un régime est indiquée, car on peut
admettre un effet additif12 ) –14 ) . La seule statine
admise en Suisse dès l’âge de 8 ans est la
pravastatine (Selipran®).
L’ézétimibe est une substance active relative-
ment récente, inhibant la résorption du cho-
lestérol au niveau des microvillosités entéro-
cytaires du grêle par blocage d’un transporteur
de stérols (NPC1L1). La diminution du taux de
cholestérol se situe autour de 25%. Son utili-
sation peut donc se justifier lors d’une hyper-
cholestérolémie familiale. En raison de l’ab-
sence d’études à long terme et du peu
d’expérience avec son utilisation chez l’enfant,
l’ézétimibe ne peut actuellement être consi-
déré que comme médicament de réserve.
Le mode d’action des fibrates n’est actuelle-
ment pas pleinement éclairci; on suppose
qu’ils stimulent l’activité de la lipoprotéine li-
pase et favorisent donc le passage intracellu-
laire des triglycérides. Les fibrates ne jouent
pas un rôle important dans le traitement des
enfants.
LDL-aphérèse, plasmaphérèse
En dernier recours la LDL-aphérèse et la plas-
maphérèse peuvent être effectuées déjà chez
l’enfant lorsqu’on n’obtient, par les autres
mesures, une stabilisation métabolique suffi-
sante.Ces procédés doivent en tout cas rapi-

23
Vol. 24 No. 4 2013 Formation continue
dement être évoqués tôt en présence d’une
hypercholestérolémie familiale homozygote.
Suivi
Au début du traitement, l’objectif du suivi
clinique est d’améliorer la compréhension des
parents et des patients, afin de renforcer la
compliance. Les patients doivent être motivés
à suivre autant que possible leur régime et, si
nécessaire, régulièrement leur médication. Le
défi est particulièrement grand lorsqu’il s’agit
de décrire la nécessité d’un traitement consé-
quent, sans éveiller des peurs exagérées, à un
enfant ou à un adolescent.
Il est indiqué d’effectuer les contrôles biochi-
miques tous les 6 mois au début, puis tous les
12 mois dès que la situation métabolique est
stabilisée. En cas de médication par des sta-
tines, les transaminases et la créatine kinase
plasmatiques doivent être contrôlés. Les
prises de sang seront effectuées de préfé-
rence à jeun, tout en sachant que le taux de
cholestérol n’augmente, que peu après un
repas contrairement à celui des triglycérides.
L’utilité de la mesure échographique de
l’épaisseur de l’intima carotidienne dépend
fortement du matériel disponible (sondes li-
néaires à très haute définition) ainsi que de
l’expérience et de la disponibilité de l’exami-
nateur. Les échographies qui ne sont pas ef-
fectuées par la même personne expérimentée
risquent d’être, dans la pratique clinique,
plutôt source d’inquiétudes.
En présence d’un taux de cholestérol très
élevé et d’une anamnèse familiale très char-
gée, des contrôles cardiologiques réguliers
(échographie cardiaque et ECG d’effort) sont
indiqués.
Pronostic
Comme nous l’avons exposé, le pronostic des
dyslipidémies dépend de nombreux facteurs
et n’est pas simplement fonction de l’amélio-
ration ou de la normalisation des paramètres
biochimiques. Il faut une fois de plus souligner
que le pronostic est favorablement influencé
par l’évitement de facteurs de risque supplé-
mentaires et par l’optimisation des conditions
de vie conjointement à l’amélioration des
paramètres biochimiques1)–3).
Résumé
Les troubles du métabolisme lipidique repré-
sentent un facteur de risque significatif pour
le développement précoce de lésions artério-
sclérosiques et doivent donc être diagnosti-
qués et traités aussi tôt que possible. Eu
égard à l’étiologie multifactorielle de l’artério-
sclérose, l’anamnèse familiale minutieuse est
importante lorsqu’il s’agit d’évaluer les
risques. En présence d’une anamnèse fami-
liale positive, des investigations ciblées (para-
mètres lipidiques) sont indiquées déjà pen-
dant la petite enfance, et même en l’absence
d’une anamnèse familiale positive un dosage
unique du cholestérol à l’adolescence est
raisonnable. Si les taux de cholestérol LDL
sont élevés, un régime modifiant l’apport en
graisses est indiqué déjà pendant la petite
enfance, associé, selon l’évolution, à un trai-
tement médicamenteux. En première inten-
tion seront utilisées les statines, du moins dès
l’âge de 8 ans. Les médicaments qui traitent
une dyslipidémie ne remplacent pas mais
accompagnent un régime.
La prise en charge des enfants avec une dys-
lipidémie devrait se faire en collaboration
avec un centre spécialisé dans le traitement
des maladies métaboliques, du moins lors de
la phase diagnostique initiale et de mise en
place du traitement.
Conclusions pour la pratique
Les auteurs de cet article estiment important
de ne pas centrer l’attention des familles
concernées uniquement sur le taux de choles-
térol, mais de considérer celui-ci comme un
facteur de risque parmi d’autres. Outre la ré-
duction d’un taux élevé de cholestérol, la
prise en compte des conditions de vie, de
l’alimentation, de l’activité physique, du poids
sont des défis tout aussi importants.
L’anamnèse familiale est la pierre angulaire
d’une évaluation globale. Lorsqu’elle est posi-
tive, des dosages ciblés des lipides plasma-
tiques sont indiqués déjà pendant la petite
enfance.
Le traitement des dyslipidémies est complexe
et inclut une intensification de l’activité phy-
sique et l’évitement de facteurs de risque
supplémentaires tout autant que le régime et
les médicaments.
Références
1) Daniels SR, Greer FR.Lipid screening and cardio-
vascular health in childhood.Pediatrics 2008; 122
(1): 198–208.
2) Peterson AL, McBride PE.A review of guidelines for
dyslipidemia in children and adolescents. Wmj
2012; 111 (6): 274–81; quiz 282.
3) Koletzko.Leitlinien zur Diagnostik und Therapie von
Hyperlipidämien bei Kindern und Jugendlichen.
Arbeitsgemeinschaft für Pädiatrische Stoffwech-
selstörungen (APS) 2007; http://www.aps-med.
de/documents/hyperlipid-22-12-2007.pdf.
4) Hovingh GK et al.Diagnosis and treatment of fami-
lial hypercholesterolaemia.Eur Heart J2013; 34
(13): 962–71.
5) MiserezAR,MullerPY.Familial defective apolipopro-
tein B-100: a mutation emerged in the mesolithic
ancestors of Celtic peoples? Atherosclerosis 2000;
148 (2): 433–6.
6) HegeleRA.Plasma lipoproteins: genetic influences
and clinical implications. Nat Rev Genet 2009; 10
(2): 109–21.
7) Brown MS, Kovanen PT, GoldsteinJL.Regulation of
plasma cholesterol by lipoprotein receptors.
Science 1981; 212 (4495): 628–35.
8) Niu DM et al.Clinical observations, molecular gene-
tic analysis, and treatment of sitosterolemia in in-
fants and children. J Inherit Metab Dis 2010; 33 (4):
437–43.
9) DurringtonP.Dyslipidaemia. Lancet 2003; 362
(9385): 717–31.
10) Wang TJ et al. Carotid intima-media thickness is
associated with premature parental coronary heart
disease: the Framingham Heart Study. Circulation
2003; 108 (5): 572–6.
11) Creider JC, Hegele RA, JoyTR.Niacin: another look
at an underutilized lipid-lowering medication. Nat
Rev Endocrinol 2012; 8 (9): 517–28.
12) Belay B, BelamarichPF, Tom-RevzonC. The use of
statins in pediatrics: knowledge base, limitations,
and future directions.Pediatrics 2007; 119 (2):
370–80.
13) Avis HJ et al.A systematic review and meta-analysis
of statin therapy in children with familial hypercho-
lesterolemia.ArteriosclerThrombVascBiol 2007; 27
(8): 18 03–10.
14) De Ferranti S, LudwigDS.Storm over Statins — The
Controversy Surrounding Pharmacologic Treat-
ment of Children, New Engl J Med 2008; 359 (13):
1309–1312.
15) De Franca E, Alves JG, HutzMH.Apolipoprotein E
polymorphism and its association with serum lipid
levels in Brazilian children.HumBiol 2004; 76 (2):
267–75.
Correspondance
Prof. Johannes Häberle
Abteilung für Stoffwechselkrankheiten
Universitäts-Kinderspital Zürich
Steinwiesstrasse 75
CH-8032 Zürich
Johannes.Haeberle@kispi.uzh.ch
Les auteurs certifient qu’aucun soutien fi-
nancier ou autre conflit d’intérêt n’est lié à
cet article.
1
/
4
100%