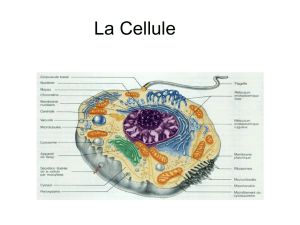
Génome et chromatine : de l`analyse multi

Génome et chromatine : de l’analyse multi-échelle des
séquences ADN à la modélisation de la réplication
chez les mammifères.
Séminaire donné par Alain Arnéodo
Laboratoire Joliot-Curie / Laboratoire de Physique de l'ENS Lyon
24 octobre 2005
Introduction
L'ADN est une macromolécule complexe en forme de double hélice qui contient de
l'information codée à travers les séquences de bases azotées A, C, T et G. Un gène est constitué
d'une alternance de séries codantes (les exons), qui contiennent l'information nécessaire à la
synthèse de protéines, et de séquences non codantes (les introns). Chez l'homme, le pourcentage
de non-codant atteint 96%. Quelle information contient ce non-codant? Comment extraire
l'information contenue dans l'ADN à partir des séquences ACGT? Cela s'apparente à un problème
de cryptographie très complexe. Pour le physicien, la méthode utilisée est de transformer d'une
manière appropriée le codage en signal exploitable et de chercher des corrélations après
traitement de celui-ci. Le signal étant constitué d'un bruit et d'une oscillation à grande échelle,
nous allons voir que :
–L'analyse du bruit nous donnera des informations sur les nucléosomes et sur les étapes
de la « compactification » de l'ADN jusqu'à la formation de chromatine;
–L'étude du signal à plus grande échelle nous renseignera sur la structure et la
dynamique de la chromatine, ainsi que sur les mécanismes de transcription et de
réplication.
I. Méthode et traitement du signal
Prenons un exemple simple de signal que l'on peut obtenir à partir d'une séquence d'ADN :
on binarise le système en attribuant la valeur +1 à A ou G et -1 à C ou T. On obtient un signal qui
s'apparente à une marche au hasard (voir figure 1). Ce signal est irrégulier dans le cas d'un exon
ou d'un intron, mais présente une invariance d'échelle dans le cas des introns. Ce résultat a mis du
temps à être accepté dans la communauté scientifique car les bioinformaticiens ont l'habitude de
chercher des invariances par translation mais pas par dilatation. Comment caractériser cette
invariance d'échelle? On utilise la technique de la variance, qui consiste à regarder comment se
comporte l'écart-type en fonction de l'échelle de longueur L. On trouve une loi de puissance :
Pour un problème de marche au hasard, en fonction de la valeur de H, on distingue
différents comportements :
–0,5 < H < 1 : Corrélation à grande échelle;
–H = 0,5 : Pas non corrélés;
–0< H < 0,5 : Anti-corrélation des pas.

Figure 1 : signal obtenu à partir d'une séquence d'ADN en « binarisant » l'information
La technique de la variance est valable uniquement pour les signaux stationnaires. Or, on
observe des fluctuations à grande échelle dans nos signaux, ce qui indique qu'ils sont non-
stationnaires. Pour remédier à cela nous devons faire appel à l'analyse en ondelettes,
généralisation de l'analyse de Fourier à chaque instant t. Cette méthode consiste à convoluer notre
signal avec un filtre de type gaussienne au carré.
En calculant l'écart-type W en prenant plusieurs valeurs de L, on peut ainsi trouver H et
conclure sur le type de corrélation.
II. Analyse du bruit : signature des nucléosomes ?
D'autres tables de codage existent, chacune servant à caractériser une propriété
particulière de la molécule d'ADN. Par exemple, des informations sur la structure peuvent être
obtenues à partir de la courbure de la double hélice, qui dépend de la séquence de chaque
nucléosome (l'axe de la double hélice étant rarement « droit »). Ou encore, les contraintes
mécaniques peuvent être étudiées via la connaissance du module d'élasticité en fonction du
séquençage. Selon le codage, on ne trouve pas la même valeur de H.
En faisant l'analyse du bruit à différentes échelles (tout en restant en deçà de 20000 paires
de bases pour pouvoir ignorer les fluctuations à grande échelle) pour des échantillons de nature
diverses, nous obtenons les résultats suivants :
Figure 2 : Valeurs de H pour un eucaryote (à gauche) et une bactérie (à droite) en fonction de L.
Les droites H=0,5 (pas de corrélation) et H=0,8 (corrélation à grande portée) sont tracées.

–Pour les cellules type eucaryote, les valeurs de H supérieures à 0,5 indiquent la
présence de corrélations à longue distance;
–Pour les bactéries (sans noyau donc sans nucléosome) on distingue deux régimes :
pour L inférieur à 200 paires de bases (ordre de grandeur de la taille d'un
chromosome) il n'y a pas de corrélation (H=0,5), pour L supérieur à 200 paires de
bases il y a des corrélations à grande distance.
Ces résultats permettent-ils de distinguer une cellule eucaryote d'une bactérie à coup sûr,
c'est à dire de mettre en évidence la présence ou non de nucléosome?
Pour valider ceci, des tests ont été réalisés à l'aveugle sur 100 virus de différents type
(eucaryote/bactérie) et le diagnostique fut bon pour 96 d'entre eux. Ces 4% d'erreurs pourraient
être acceptable pour un physicien, mais le biologiste cherche à comprendre pourquoi ces quatre
virus « eucaryote » ont été diagnostiqué « bactérie » avec la méthode de la variance. En y
regardant de plus près, il se trouve que ces quatre exceptions sont des « pox virus » qui ont la
particularité de se répliquer dans le cytoplasme et non dans le noyaux, comme c'est
habituellement le cas. Le processus de réplication ne mettant pas en jeu de nucléosome, ces pox
virus ont donc été diagnostiqués comme étant de type bactérie.
La méthode de la variance permet donc de différencier rapidement les virus qui se
répliquent dans le noyau de ceux qui se répliquent dans le cytoplasme, ce qui n'avait jamais pu
être mis en évidence auparavant.
En outre, le régime de corrélations à longue portée induit la formation aisée de boucles de
petite taille (fait expliqué par la modélisation de la formation de ces boucles en physique
statistique), ce qui explique la structure de la chromatine. En effet, le minimum d'energie libre
nécessaire à la formation d'une boucle de taille L diminue avec H.
III. Analyse à grande échelle : mécanisme de transcription?
En se plaçant à une échelle supérieure à 20kb, après lissage du bruit on observe des
oscillations. Deux « bosses » apparaissent : une vers 100kb, taille caractéristique des boucles de
chromatine et une à 400kb, taille moyenne pour le domaine de réplication chez les mammifères.
Cherchons un codage adapté à l'étude du mécanisme de transcription: nous remarquons que la
transcription et la réplication nécessitent l'ouverture de la double hélice et désymétrisent les brins.
Il nous faut donc un codage qui rende compte de la symétrie des brins d'ADN.
Sur une molécule d'ADN, les bases azotées étaient associées par paires AT (ou TA) et CG
(ou GC), le nombre de bases A (noté [A]) est égal au nombre de bases B (noté [B]), de même
[G]=[C]. Quand cette symétrie est brisée, on peut la quantifier en introduisant les quantités SCG et
SAT .
A l'échelle du génome, ces quantités sont nulles, mais à l'échelle d'un million de paires de
bases des fluctuations sont visibles. Ce sont elles que nous allons étudier.
A petite échelle, nous retrouvons dans le bruit des fluctuations d'ordre de grandeur de
100kb et 400kb. A grande échelle, SCG et SAT sont positifs sur quelques millions de paires de base,
puis négatifs sur la même longueur, et ainsi de suite. Comment relier ce comportement aux
caractéristiques physique de la molécule?
Dans le mécanisme de réplication, de l'origine au terminus il y a deux sens possibles : le
chemin rouge ou le chemin bleu (voir page suivante).

Figure 3
Sur la figure 3 ci-dessus représentant le signal de biais (SCG), on peut noter les deux
régimes différents: sur 2 Mb les gènes anti-sens, en bleu, dominent puis ce sont les gènes sens,
en rouge. Les gènes anti-sens étant en bas des oscillations et les genes sens en haut, on observe
un saut ascendant puis descendant tous les 2Mb.
Que pourraient représenter ces sauts? Le génome humain comportant environ quatre
milliards de paires de base et un domaine de réplication étant d'environ 400kb, il existe des
milliers de sites de réplications. Or, nous n'en connaissions que neuf au moment de ces
expériences. En se plaçant à chacun de ces sites connus, nous remarquons qu'il coïncide avec un
de ces sauts positifs dans le signal bruité. Ceci n'est pas qu'une pure coïncidence, car après
vérifications dans le génome de la souris, du singe, du rat etc. , nous retrouvons la même
corrélation entre origine de réplication et saut positif. Cette méthode permet ainsi de prédire plus
de 2000 origines de réplication.
IV. Résultats
On rappelle que l'analyse du bruit et des corrélations à longue portée permet de
différencier les processus de réplication mettant en jeu on non des nucléosomes et permet
également d'expliquer et de quantifier les sctructures en boucle de la chromatine.
Un résultat important de l'analyse à grande échelle est qu'il y a un sens, une parité dans le
génome, qui régit son organisation : les gènes sens se répliquent vers la droite, les gènes anti-
sens vers la gauche.
On remarque également que l'origine est toujours à la même position dans le génome mais
que la position du terminus varie. Comment comprendre la coordination et la régulation pour que
le mécanisme se déroule sans encombres? C'est la structure et la dynamique de la chromatine qui
s'adapte à cette situation en adaptant une forme en « Rosette » afin de laisser le plus de place
aux protéines.
1
/
4
100%