La réparation de l`ADN par la recombinaison homologue et le

La réparation de l'ADN par la recombinaison homologue
et le développement de molécules anticancéreuses
Thèse
Joris Pauty
Doctorat en biologie cellulaire et moléculaire
Philosophiae Doctor (Ph.D.)
Québec, Canada
© Joris Pauty, 2015


iii
Résumé
Le cancer est une cause majeure de décès dans le monde. Il est à présent établi que les mutations de
l'information génétique des cellules initient et participent à son développement, et que certaines mutations
transmises au sein des familles prédisposent à son apparition. C'est le cas notamment des mutations des gènes
BRCA1 et BRCA2 qui prédisposent aux cancers du sein et de l'ovaire. Les protéines produites par ces gènes
sont directement impliquées dans la protection de l'information génétique puisqu'elles participent à la réparation
des cassures se produisant dans le support de cette information : l'ADN. L'ADN peut être endommagé par
diverses lésions mais les plus déstabilisatrices de l'information génétique sont les cassures double-brin. Afin de
protéger son génome, la cellule possède de nombreux mécanismes de réparation dont la recombinaison
homologue qui permet une réparation fidèle, c'est-à-dire sans perte ou modification de l'information génétique,
permettant ainsi de prévenir l'apparition du cancer. La recombinaison homologue repose principalement sur
l'activité de la protéine RAD51 qui nécessite l'utilisation des médiateurs BRCA2 et PALB2. Tout comme les gènes
BRCA1 et BRCA2, PALB2 est un gène suppresseur de tumeur et ses mutations ont été associées avec une
susceptibilité aux cancers du sein, de l'ovaire et du pancréas. En plus de la chirurgie, le traitement de ces cancers
implique la radiothérapie et la chimiothérapie. Celles-ci font l'objet d'intenses recherches afin de proposer de
nouveaux traitements plus efficaces avec moins d'effets secondaires. De nouvelles stratégies
chimiothérapeutiques ont notamment émergé et on s'oriente à présent vers le développement de traitements
personnalisés qui sont basés sur une meilleure connaissance des spécificités moléculaires des tumeurs.
Les travaux présentés dans cette thèse apportent de nouvelles informations concernant le rôle de
PALB2 dans la protection du génome lors du stress réplicatif et sur la régulation de ses fonctions par le contrôle
de sa localisation cellulaire. Plus précisément, nous montrons que PALB2 et BRCA2 permettent de maintenir la
Polymérase η au niveau des fourches de réplication bloquées et stimulent son activité de synthèse de l'ADN pour
réinitier la réplication. Grâce à l'analyse de mutations germinales identifiées dans des cancers du sein et de
l'ovaire, nous révélons la présence d'une séquence d'export nucléaire qui provoque l'exclusion de PALB2 du
noyau vers le cytoplasme. Enfin, nous rapportons le développement d'une nouvelle molécule
chimiothérapeutique, SFOM-0046, qui provoque des cassures double-brin de l'ADN en induisant un stress
réplicatif et qui potentialise les effets de l'UCN-01, une molécule qui a été étudiée en clinique. Nous proposons
l'utilisation de cette nouvelle molécule comme agent d'amélioration de thérapies ciblées existantes ou pour le
développement de nouvelles thérapies anticancéreuses personnalisées.


v
Table des matières
Résumé ................................................................................................................ iii
Table des matières ................................................................................................ v
Liste des abréviations ........................................................................................... ix
Liste des tableaux ............................................................................................... xiii
Liste des figures ................................................................................................... xv
Remerciements .................................................................................................. xix
Avant-Propos ................................................................................................... xxiii
Chapitre 1 ..................................................... 1
Chapitre 1. Introduction ....................................................................................... 3
1.1. La réponse aux cassures double-brin de l'ADN ................................................................... 4
1.1.1. La surveillance du génome: les points de contrôle et la détection des cassures ............. 4
1.1.1.1. Les acteurs moléculaires des points de contrôle ........................................................ 6
1.1.1.2. Le point de contrôle G1/S .......................................................................................... 7
1.1.1.3. Le point de contrôle intra-S ........................................................................................ 8
1.1.1.4. Le point de contrôle G2/M ......................................................................................... 8
1.1.1.5. Le point de contrôle de restauration de la réplication ............................................. 10
1.1.1.6. La détection et la signalisation d'une cassure double-brin ...................................... 10
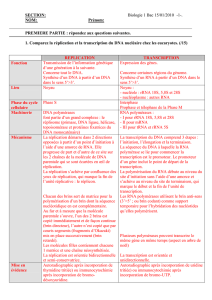
1.1.2. La réparation des cassures double-brin .......................................................................... 13
1.1.2.1. La jonction d'extrémités non-homologues : les voies classique et alternative ........ 14
1.1.2.2. La recombinaison homologue .................................................................................. 17
1.1.2.3. L'appariement des extrémités protubérantes simple-brin ....................................... 20
1.1.3. Les maladies associées à un défaut de la réparation de l'ADN ....................................... 21
1.2. Focus sur certains acteurs de la réparation de l'ADN ....................................................... 23
1.2.1. La recombinase RAD51 ................................................................................................... 23
1.2.2. BRCA1 et BRCA2 .............................................................................................................. 24
1.2.2.1. BRCA1 ...................................................................................................................... 24
1.2.2.2. BRCA2 ...................................................................................................................... 24
1.2.3. PALB2 et les ADN polymérases ....................................................................................... 26
1.2.3.1. Le suppresseur de tumeur PALB2 ............................................................................. 26
1.2.3.2. Les ADN polymérases dans la réparation des dommages à l'ADN .......................... 26
1.3. Les stratégies thérapeutiques contre le cancer utilisant les cassures double-brin ............ 28
1.3.1. Les stratégies thérapeutiques ......................................................................................... 28
1.3.1.1. Les stratégies existantes et leurs perspectives de développement .......................... 28
1.3.1.2. Le principe de létalité synthétique ........................................................................... 29
1.3.2. Les agents thérapeutiques conventionnels .................................................................... 31
1.3.2.1. La radiothérapie ...................................................................................................... 31
1.3.2.2. La chimiothérapie .................................................................................................... 31
1.3.2.3. Les inhibiteurs de topoisomérase ............................................................................ 31
1.3.3. Les nouvelles classes d'agents thérapeutiques en développement ............................... 34
1.3.3.1. Les inhibiteurs de PARP ............................................................................................ 34
1.3.3.2. Les inhibiteurs des senseurs des cassures double-brin et du cycle cellulaire ........... 35
1.3.3.3. Les inhibiteurs du NHEJ ............................................................................................ 36
1.3.3.4. Les inhibiteurs de la Recombinaison Homologue .................................................... 37
1.3.4. Les limites et perspectives des traitements .................................................................... 39
Objectifs ............................................................................................................. 41
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
192
193
194
195
196
197
198
199
200
201
202
203
204
205
206
207
208
209
210
211
212
213
214
215
216
217
218
219
220
221
222
223
224
225
226
227
228
229
230
231
232
233
234
235
236
237
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
192
193
194
195
196
197
198
199
200
201
202
203
204
205
206
207
208
209
210
211
212
213
214
215
216
217
218
219
220
221
222
223
224
225
226
227
228
229
230
231
232
233
234
235
236
237
1
/
237
100%