II. Métabolisme des acides aminés

Noémie VAUCHER
Kevin CHEVALIER 1
METABOLISME DE L'AZOTE
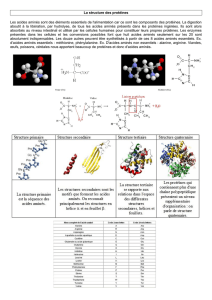
On a ici les voies amenant les acides aminés qui proviennent des protéines endogènes et des
protéines alimentaires. Ceci donne les acides aminés circulants qui sont utilisés pour être dégradés
et forment de l'énergie. Mais ils servent aussi à la constitution de protéines, de neurotransmetteurs,
d'hormones, etc…
I. Apports azotés
Les apports azotés en acides aminés sont constitués par des protéines :
Exogènes (75 g/jour)
Endogènes (225 g/jour), provenant du renouvellement de 600 g de muscles et 200 g d'os.
A. Protéines alimentaires
1. Apport alimentaire
L'apport alimentaire représente 10 à 15% de la ration calorique car leur valeur énergétique est
faible (4kcal/g).
L'organisme ne dispose d'aucune réserve protéique contrairement au glucose et aux acides gras : Le
bilan azoté est nul.
En cas de force majeure, les protéines musculaires sont utilisées.
La valeur nutritive d'une protéine dépend de sa composition en acides aminés. La protéine idéale est
l'ovalbumine (blanc d'œuf) qui apporte les acides aminés indispensables dans les proportions idéales
pour l'homme.
Il est donc recommandé de varier les apports en protéines.

Noémie VAUCHER
Kevin CHEVALIER 2
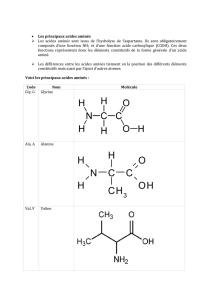
2. Acides aminés essentiels (9 acides aminés)
Ce sont des acides aminés ne sont pas synthétisables par l'organisme : Histidine (His), Isoleucine
(Ileu), Phénylalanine (Phe), Valine (Val), Méthionine (Met), Leucine (Leu) Tryprotphane (Try), Lysine
(Lys) Thréonine (Thr).
Phrase mnémotechnique :"L'hystérique Iseult fait vachement marcher le très lyrique Tristan".
3. Origine
Les protéines animales ont pour origine : les viandes, les poissons, les œufs, les laitages. L'origine
végétale est constituée par le pain (gluten), les céréales, les légumineuses.
Les végétaliens (à la différence des végétariens, ils ne mangent pas d'œufs ni de produits laitiers)
peuvent manquer d'acides aminés essentiels lors de la croissance.
4. Acides aminés non indispensables
Ils sont synthétisés à partir :
D'autres acides aminés. Exemple : La sérine (Ser) donne la Glycine (Gly) et la Cystéine (Cys)
D'intermédiaires de la glycolyse et du cycle de Krebs (et donc du glucose)
B. Protéines endogènes
1. Localisation des synthèses
Le renouvellement des protéines est plus intense dans le foie, les muscles, l'intestin et la peau.
Le foie est le site le plus important de synthèse des protéines. Il synthétise :
L'albumine
Les parties protéiques des lipoprotéines (ApoB100 des LDL…)
Les protéines de transport d'hormones et de vitamines :
CBG: Cortisol Blinding Protein
TBG: Thyroxine Blinding Protein

Noémie VAUCHER
Kevin CHEVALIER 3
2. Régulation de la synthèse endogène
La régulation de la synthèse des protéines endogène est hormonale :
En période alimentaire : La synthèse domine sous l'effet des acides aminés exogènes et de
l'insuline, hormone favorisant la mise en réserve.
En période de jeûne : Le catabolisme domine, sous l'effet des hormones du stress : le
cortisol et l'adrénaline.
En cas d'apport protéique insuffisant, on parle de marasme ou Kwashiorkor.
C. Transport plasmatique
Les acides aminés les plus abondants dans le sang sont l'alanine (Ala) et la glutamine (Gln), produits
essentiellement par les muscles (45% du total).
Le foie capte 75% des acides aminés pour le renouvellement de ses protéines.
Les muscles captent surtout les acides aminés ramifiés : La Valine (Val), Isoleucine (Ileu), la leucine
(leu).
Les reins captent la glutamine (Gln) pour la synthèse des urées qui est la forme d'élimination de
l'azote.
II. Métabolisme des acides aminés
A. Libération du squelette carboné par désamination
La désamination, permet de récupérer le squelette carboné et une molécule d'azote (NH3).
Elle est oxydative car elle donne un acide α cétonique.

Noémie VAUCHER
Kevin CHEVALIER 4
La désamination n'est pas directe mais se fait en deux étapes successives :
La première étape est le transfert du groupe –NH2 sur un accepteur : α-céto glutarate qui
devient Glu par transamination
La seconde étape est la désamination, oxydative, de Glu qui redonne l' α-céto glutarate et un
azote. Cela permet de récupérer un NADH et donc de créer 3 ATP.
Les transaminases les plus abondantes sont l'Alat (Alanine Amino Transférase) et l'Asat (Aspartate
Amino Transférase).
L'alanine, donne, grâce à l'ALAT et l'αCG du pyruvate et du glucose. Cette réaction est
réversible.
Pour l'aspartate, on aura la même voie, sauf qu'on aura de l'oxalo acétate.
Ces transaminases sont abondantes, surtout dans le foie. Leur relargage dans le sérum signe une
cytolyse hépatique (hépatite, cirrhose).
B. Destinée du squelette carboné
On distingue trois groupes d'acides aminés selon la destinée du squelette carboné :
GROUPE 1
GROUPE 2
GROUPE 3
Particularités
Acides aminés à 3 carbones.
C'est le groupe le plus abondant.
Acides
aminés
Ala, Thr, Gly, Ser, Cys
Glu, Gln, Arg, His, Pro, Ile, Met, Val,
Phe, Tyr, Asp, Asn
Leu, Lys, Trp, Phe, Tyr
Destinée du
squelette
carboné
Pyruvate
Il donne des intermédiaires du cycle
de Krebs. La fin du cycle de Krebs
donnera l'oxalo-acétate qui permet de
produire du glucose ou de l'énergie.
Il donne l'acétoacétyl-CoA qui est au
début de la cétogenèse qui donne les
corps cétoniques.
Action(s)
Glycogènes : Ils redonnent
du glucose par la
gluconéogenèse.
Fournit de l’énergie par
une dégradation
Glycogènes : Ils redonnent du
glucose par la gluconéogenèse.
Fournit de l’énergie par une
dégradation
Cétogènes : L'acétoacyl-CoA conduit
aux corps cétoniques donc les acides
du groupe 3 sont dits cétogènes.
Le choix entre les groupes 1 et 2 pour la synthèse de glucose dépend de l'état énergétique et
hormonal de la cellule.

Noémie VAUCHER
Kevin CHEVALIER 5
C. Synthèse d'amine par décarboxylation
La décarboxylation ne concerne que certains acides aminés et certaines cellules (elle n’est pas
systématique) : les cellules nerveuses, les cellules immunitaires, … La décarboxylation donne des
amines à activités biologiques :
1. Réaction générale
L'acide aminé perd son CO2 par décarboxylation pour donner une amine. Une fois son rôle
biologique achevé, il faudra l'inactiver et l'éliminer.
L'inactivation se fait par une désamination oxydative (½ O2) donnant un aldéhyde.
L'oxydation ne s'arrête pas au stade de l'aldéhyde mais continue pour oxyder l'aldéhyde en acide. On
ajoute encore ½ O2 pour avoir un R-COOH. C'est sous forme d'acide que l'amine sera éliminée par les
reins.
2. Acides aminés spécifiques
L'histidine donne l'histamine, médiateur du choc allergique.
Le 5-hydroxy-tryptophane donne la sérotonine, neuromédiateur du bien-être.
Le DOPA (Di-hydroxy Phényl Alanine) donne la dopamine, neuromédiateur du bien être et
de la mémoire.
L'enzyme de dégradation des amines du cerveau est la Mono-Amine-Oxydase ou MAO. On utilise
des inhibiteurs de cette enzyme : les IMAO comme antidépresseurs.
Le glutamate donne le GABA (Acide Gamma Amino Butyrique) neuromédiateur inhibiteur
du système nerveux central (médiateur du sommeil).
Création de l'amine
Inactivation et élimination
 6
7
8
9
10
11
6
7
8
9
10
11
1
/
11
100%