Myopathie de Duchenne : Symptômes, Génétique & Thérapie

Myopathie de Duchenne
Document 1 : les symptômes (http://www.caducee.net/DossierSpecialises/genetique/myopathie-
duchenne.asp)
La myopahtie de Duchenne (DMD : Dystrophie musculaire de Duchenne) se caractérise par une
dégénérescence musculaire qui conduit à la paralysie progressive du malade, condamné au fauteuil roulant
vers l’âge de 10 ans. Elle est due à des mutations récessives du gène codant la dystrophine, protéine de la
membrane plasmique des cellules musculaires. Ce gène est situé sur le chromosome sexuel X : la maladie est
dite gonosomale. Elle affecte essentiellement les garçons, avec la fréquence de 1 sur 3500 naissances dans la
population française. Un enfant atteint de myopathie de Duchenne présente en général peu de signes de la
maladie avant l'âge de 3 ans. Il marche parfois tard, tombe assez souvent et se relève difficilement. Les autres
symptômes sont :
- Hypertrophie des mollets et atrophie d'autres muscles
- une faiblesse musculaire progressive des membres et du tronc : la marche deviennent impossibles
vers 10-12 ans et l'utilisation des membres supérieurs se limite progressivement.
- Une scoliose souvent grave se développe mais le plus souvent après la perte de la marche.
- L'atteinte des muscles respiratoires rend l'enfant sensible aux infections pulmonaires.
- Des troubles cardiaques sont fréquents (trouble de la conduction, du rythme).
- Un retard intellectuel est possible mais ne concerne qu'une partie des enfants atteints par la maladie
- Un enraidissement des articulations: du à la formation de contractures musculaires au niveau des
articulations. Ces contractures touchent d'abord les chevilles (le tendon d'Achille), puis les hanches et les
genoux, enfin les articulations des bras. Cette complication est liée à l'atrophie des muscles qui, ne servant
plus, finissent par se rétracter.
Grâce à une prise en charge globale et adaptée l'enfant atteint le plus souvent l'âge de 20 - 30 ans.
Document 2 : Phénotype cellulaire

Document 3 : Phénotype moléculaire
La dystrophine est une protéine de la membrane des cellules musculaires. Elle est
fondamentale pour la contraction musculaire car c’est elle qui ancre les fibrilles musculaires (actine
et myosine) qui ont la capacité de se raccourcir. La myopathie de Duchenne mutations est liée à une
mutation qui conduit à la production d’une protéine tronquée. Dans ce cas, la protéine ne sera pas
exprimée et elle sera tout de suite dégradée par la cellule.
En absence de dystrophine, les myofibrilles contractiles ne s’ancrent plus sur les membranes
des cellules musculaires et sont désorganisées. La contraction musculaire devient alors très peu
efficace et le tissu musculaire se désorganise.
Document 4 : Phénotype moléculaire
Voici un extrait des séquences de nucléotides des gènes de la dystrophine (nucléotide 327 à
342) chez un individu sain et chez un individu malade. Ces nucléotides correspondant aux acides
aminés 109 à 114 de la dystrophine.
Séquence individu sain : ------- CCAAACTAGACCTTATAT -----------------
Séquence individu malade : ------- CCAAACTAAACCTTATAT -----------------
Document 5 : La transmission de la myopathie de Duchenne

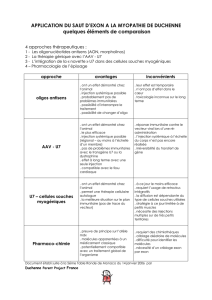
Document 6 : La thérapie génique par « exon skipping » (http://www.futura-
sciences.com/fr/news/t/vie-1/d/la-myopathie-de-duchenne-corrigee-chez-la-souris_4738/)
Mise au point d'une technique
innovante de thérapie génique : le
saut d'exon. Une équipe de
chercheurs de Généthon, le
laboratoire créé et financé par
l'Association Française contre les
Myopathies (AFM) grâce aux dons du
Téléthon, a réussi à réparer les
muscles de souris modèles de la
myopathie de Duchenne, grâce à une
technique de thérapie génique
appelée « saut d'exon » (exon
skipping).
Pour ce faire, ils ont introduit dans la cellule, grâce à un vecteur AAV (Adeno Associated Virus),
une molécule appropriée pour que l'épissage ignore l'exon défectueux (chez ces souris, il s'agit de l'exon 23). La
molécule utilisée est un petit ARN (Acide RiboNucléique) du noyau cellulaire appelé U7, pouvant être modifié
pour intervenir au moment de l'épissage. U7 va masquer l'exon défectueux et ainsi rétablir le cadre de lecture
dans la cellule. Le couple AAV-U7 a été injecté dans le muscle d'une patte de souris adultes (âgées de 8
semaines) et, pour un second groupe de souris, par perfusion intra-artérielle. Dans les deux groupes, la
dystrophine, absente des cellules musculaires, a été détectée à partir de 4 semaines après l'injection dans la
plupart des fibres du muscle et les souris traitées ont montré des performances musculaires équivalentes aux
souris saines. Depuis plus de six mois, le niveau d'expression de la dystrophine reste stable chez ces souris.
Les chercheurs ont poursuivi des études pré-cliniques afin de préparer un essai de phase 1 sur l'Homme
d'ici 2007. Ils se concentreront dans un premier temps sur l'exon 51. En ciblant seulement 6 exons, cette
technique pourrait concerner, chez l'Homme, 85% des délétions du gène de la dystrophine (base de données
de JC Kaplan, Institut Cochin).
1
/
3
100%