INFORMATION : PHRC NATIONAL APECED 2009

INFORMATION : PHRC NATIONAL APECED 2009-2011
Evaluation génotypique, phénotypique et pronostique du syndrome
APECED: étude multicentrique nationale" »
INVESTIGATEURS-COORDONNATEURS
E PROUST-LEMOINE, JL WEMEAU (CHU Lille)
COLLABORATEURS SCIENTIFIQUES
D LEFRANC, S DUBUCQUOI, L PRIN (EA 2686, Laboratoire d’Immunologie, CHU de Lille),
JC CAREL (Service de Pédiatrie, Hôpital Robert Debré, Paris), H LEFEBVRE (Service
d’Endocrinologie, CHU Rouen), P SAUGIER-VEBER (INSERM U614, Laboratoire de
Génétique Moléculaire, Rouen), R MALLONE (INSERM U561, Hôpital St Vincent de Paul,
Paris), N FABIEN (Laboratoire d’Immunologie, CHU Lyon)
ATTACHES DE RECHERCHE CLINIQUE
ARC promoteur : C PIATEK, ARC investigateur : M BOIS (CHU Lille)
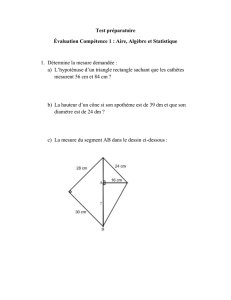
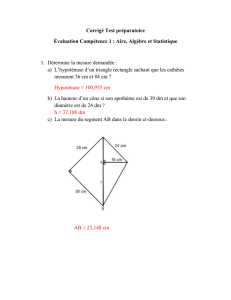
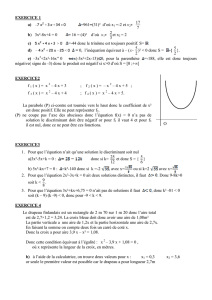
Le syndrome APECED (Auto-immune Poly-endocrinopathie Candidiasis Ectodermal
Dystrophy) ou poly-endocrinopathie auto-immune de type 1 (PEA1) est une pathologie rare,
de transmission autosomique récessive, liée aux mutations du gène AIRE. Initialement
décrite par Whitaker, cette pathologie associe des manifestations auto-immunes spécifiques
d’organe, endocriniennes et non endocriniennes, une candidose cutanéo-muqueuse
chronique et une dystrophie ectodermique.
Le mécanisme physiopathologique de cette pathologie s’avère exemplaire. En effet, le gène
AIRE, exprimé dans le thymus, les ganglions et la rate, y joue un rôle primordial dans
l’établissement de la tolérance immune, permettant la délétion clonale de populations
lymphocytaires T autoréactives vis à vis d’antigènes du soi. Dans cette pathologie, mieux
comprise grâce au modèle murin Aire -/-, cette sélection négative n’a pas lieu, les clones
lymphocytaires T auto-réactifs non délétés sont à l’origine de pathologies auto-immunes
tissu-spécifiques. Alors que le mécanisme des candidoses demeurait méconnu; deux
équipes ont récemment mis en évidence la présence d’anticorps neutralisants dirigés
contre des cytokines de type Th17, impliquées dans la défense contre les infections
fungiques, chez ces patients. Ainsi, les réponses cytokiniques de type IL-17F et IL-22 au
Candida sont diminuées, ce qui pourrait être en lien avec la survenue de candidoses chez
ces patients.
La PEA1 débute en général dans l’enfance, associant une candidose cutanéo-muqueuse

chronique, une poly-endocrinopathie auto-immune (hypoparathyroïdie, insuffisance
surrénalienne,…), des pathologies auto-immunes non endocriniennes (digestives,
cutanées, …) et des anomalies ectodermiques (hypoplasie de l’émail dentaire, kérato-
conjonctivite, dystrophie unguéale,…). Toutefois, la présentation clinique demeure variable
d’un patient à l’autre. Certains patients développent des formes atypiques, ou pauci-
symptomatiques, dont le diagnostic peut être méconnu. D’autres présentent des atteintes
graves, menaçant le pronostic vital (carcinome des muqueuses orales ou œsophagiennes,
sepsis sévère, hépatite fulminante, atteintes rénale ou bronchique). Dans les atteintes auto-
immunes sévères, un recours aux immunosuppresseurs, voire à la transplantation hépatique
ou rénale, peut s’avérer nécessaire.
Du fait de son origine monogénique, l’étude de cette pathologie a beaucoup contribué à la
compréhension des phénomènes de tolérance immune centrale, à la définition des sites
antigéniques potentiels des tissus périphériques, et à la caractérisation de nouveaux auto-
anticorps. La gravité de la maladie justifie sa meilleure reconnaissance, la meilleure
définition de ses caractéristiques phénotypiques, génotypiques et de son évolutivité dans le
cadre d'évaluations multicentriques.
Dans ce contexte, une étude nationale, faisant suite à une étude Inter-régionale réalisée
dans le Nord-Ouest de la France, a été acceptée dans le cadre d’un PHRC national. Nous
en attendons une description précise épidémiologique, clinique, immunologique et génétique
du syndrome APECED, à partir d’un recensement national. Les éventuelles spécificités
régionales du syndrome seront analysées, notamment en comparaison des informations
obtenues dans les grandes séries mondiales. Nous souhaitons favoriser sa connaissance et
son diagnostic par les praticiens, notamment dans ses formes atypiques et pauci-
symptomatiques. Cette analyse clinique et biologique devrait permettre de dégager des
facteurs pronostiques, et d’intensifier la prise en charge des formes engageant le pronostic
vital. Sur la base des informations obtenues, pourront être définies les situations justifiant
l’évaluation prospective randomisée de médications immunomodulatrices récemment
envisagées dans cette pathologie.
Références bibliographiques
Aaltonen J, et al, An autosomal locus causing autoimmune disease: autoimmune polyglandular disease type I
assigned to chromosome 21. Nat Genet. 1994;8:83-7.
Consortium, An autoimmune disease, APECED, caused by mutations in a novel gene featuring two PHD-type
zinc-finger domains. The Finnish-German APECED Consortium. Autoimmune Polyendocrinopathy-Candidiasis-
Ectodermal Dystrophy. Nat Genet. 1997;17:399-403.
Nagamine K, et al, Positional cloning of the APECED gene. Nat Genet. 1997;17:393-8.
Anderson MS, Venanzi ES, Klein L, et al Projection of an immunological self shadow within the thymus by the aire
protein. Science 2002;298:1395-1401.

Liston A, et al: Aire regulates negative selection of organ-specific T cells. Nat Immunol 2003;4:350-4.
Gardner JM, et al, Deletional tolerance mediated by extrathymic Aire-expressing cells. Science 2008;321:843-7.
Kisand K, et al, Chronic mucocutaneous candidiasis in APECED or thymoma patients correlates with
autoimmunity to Th17-associated cytokines. J Exp Med, 2010 207: 299-308.
Puel A, et al, Autoantibodies against IL-17A, IL-17F, and IL-22 in patients with chronic mucocutaneous
candidiasis and autoimmune polyendocrine syndrome type I. J Exp Med, 2010; 207:291-7.
Alimohammadi M, et al, Pulmonary autoimmunity as a feature of autoimmune polyendocrine syndrome type 1 and
identification of KCNRG as a bronchial autoantigen. Proc Natl Acad Sci U S A 2009;106:4396-4401.
Perheentupa J, Autoimmune Polyendocrinopathy-Candidiasis-Ectodermal Dystrophy. J Clin Endocrinol Metab
2006;91:2843-2850.
Husebye ES, et al: Clinical manifestations and management of patients with autoimmune polyendocrine
syndrome type I. J Intern Med 2009;265:514-529.
Proust-Lemoine E, et al, Autoimmune Polyendocrine Syndrome Type 1 in North-Western France: AIRE Gene
Mutation Specificities and Severe Forms Needing Immunosuppressive Therapies. Horm Res Paediatr
2010;74:275–284.
Pour obtenir des informations complémentaires, vous pouvez contacter :
E. Proust-Lemoine (e.proustlemoine@yahoo.fr)
J. L. Wémeau (jl-wémeau@chru-lille.fr)
1
/
3
100%