Cours 8 Demence et Demences

Cours n°8 : neurologie 18/05/2010
Démence et Démences…
Introduction
Démence, définition : Troubles cognitifs d’évolution progressive évoluant depuis plus de 6
mois avec troubles de l’adaptation socio-professionnelle et familiale
Etiologies
Neurodégénérative, la plus fréquente
Autres étiologies :
o Infectieuses : HIV, syphilis
o Creutzfeldt Jakob
o Métaboliques : carences vitaminiques (PP, B12, folates),
o dysthyroidie
o Tumorales (tumeur du système nerveux central)
o Hydrocéphalie à pression normale
o Vasculaires
Dépression du sujet âgé
Concept de démence corticale et sous-corticale
• Démence corticale
– Maladie d’Alzheimer +++
– Dégénerescences fronto-temporales
– Demence frontale
– Aphasie primaire progressive
– Démence sémantique
• Déménce sous-corticale
– Maladie de Steele-Richardson (PSP)
– Maladie de Parkinson
– Maladie de Huntington
• Cas particuliers:
– Démence à corps de lewy diffus
– Dégénérescence cortico-basale
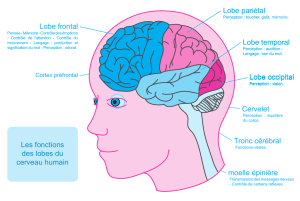
Fonctions cognitives
• Supportées par des régions corticales connectées entre elles et avec des structures
profondes du système nerveux central
• Schématiquement, on distingue
– les fonctions exécutives : lobe pré-frontal
Flexibilité mentale
Planification
Mémoire de travail

– Les fonctions mnésiques et de langage
Mémoire : hippocampe
Langage : lobe temporal externe
– Les fonctions instrumentales : lobe pariétal
Praxies gestuelles
Gnosies
Démences corticales
• Maladie d’Alzheimer
– Signes cliniques:
• Plaintes mnésiques initiales
• Phase d’état = Amnésie, apraxie, aphasie , agnosie (Syndrome des 4 A)
• Myoclonies, syndrome parkinsonien, crises d’épilepsie dans un délai
variable (en moyenne > 5 ans )
• Décès apres 8 à 10 ans d’évolution
– Diagnostic clinique de probabilité (critères DSMIV)
• Examens complémentaires pour éliminer autre cause
– Diagnostic de certitude anatomopathologique
– Traitement
• Prise en charge globale: psychologue, stimulation cognitive, assistante
sociale
• Ttt med: anticholinestérasiques
Physiopathologie
• Atteinte initialement hippocampique puis diffusion des lésions à l’ensemble du cortex,
atteinte des noyaux Nad du tronc et du noyau cholinergique basal de Meynert
• 2 types de lésions:
– plaques séniles: formations extracellulaires, centre amyloïde = peptide A4,
périphérie = neurones dystrophiques
– DNF: lésions intra neuronales faites de paires de filaments hélicoïdaux dont le
composant principal = protéine tau anormalement phosphorylée et glycosylée
Diagnostic
• Clinique:
– Plainte mnésique
– Evaluation cognitive
• Simple au lit du malade
– MMS, BREF, tests des 5 mots, test de l’horloge
• Evaluation neuropsychologique
– Grober et Buchke
• Bilan paraclinique:
– Biologique
– Imagerie
– Morphologique/scintigraphie
– PL avec étude des biomarqueurs (MCI+++)

Démences sous-corticales
• Par disconnexion entre le lobe frontal et les ganglions de la base (structures profondes
du système nerveux central). Atteinte soit des faisceaux de connexion (substance
blanche) soit des ganglions de la base eux-mêmes
Syndrome sous-cortico-frontal
• Troubles de l’attention et de la concentration
• Troubles mnésiques mais sensibles aux indiçages
• Troubles du comportement :
Apathie, apragmatisme
Désinhibition, hyperoralité
Comportement d’utilisation, d’imitation,
collectionisme
Libération de réflexes archaïques (grasping reflexe, sucking)
Maladies à Prion ou encéphalopathies subaiguës spongiformes transmissibles
Introduction
• Maladie à expression neurologique principale
• Agent transmissible non conventionnel PrPsc=protéine prion anormale
• Décrite chez
– L’homme
• Creutzfeldt-Jacob
• Insomnie fatale familiale
• Sd de Gerstmann-Straussler-Scheinker
• Kuru
– L’animal
• Ovin (tremblante du mouton, 1732)
• Bovins (vaches folles, 1986)
La protéine prion
• Présente à l’état normal sur la membrane des neurones
• Agent pathogène issue d’une transformation spatiale: PrP PrPsc (scrapie)
• Transconfiguration secondaire de proche en proche
• Proteine anormale résistante aux agents chimiques et thermiques conventionnels
Maladie de Creutzfeld-Jacob (CJ)
• Décrite en 1920
• Taux de mortalité: 0.8 à 1.4 par millions d’habitant et par an
• 4 formes cliniques
– CJ sporadique
– CJ familial
– CJ iatrogène
– CJnv
Anatomopathologie
A Spongiose

B dépôts focaux de PrPsc qui prédominent à la jonction de la couche granulaire et de
la couche des cellules de Purkinje (marquage immunohistochimique de la PrP à l’aide
de l’anticorps 3F4
C gliose astrocytaire
1
/
4
100%