Echelle Cellulaire

1
Emérentienne Michelin
Dorine Versavaud
Camille Simonet
Maladie Génétiques

2
Plan
En quoi certaines maladies génétiques
rares restent-elles des énigmes pour la
médecine d’aujourd’hui ?
I)Les différentes échelles de 3 maladies génétiques rares
a)Progéria
b)Huntington
c)Enfants de la lune
II) Difficultés et obstacles des recherches médicales
a) Au niveau financier
b) Les technique et les limites de la médecine actuelle
c) Les problèmes éthiques
III) Espoir de guérison
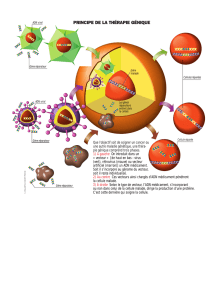
a) La thérapie cellulaire
b) La thérapie génique
c) Une avancée dans le temps
Conclusion
I

3
)Les différentes échelles de trois maladies génétiques rares
Nous avons choisi de présenter ces trois maladies sous trois échelles différentes. L’échelle
macroscopique qui décrit les symptômes de la maladie, l’échelle cellulaire qui s’intéresse au
gène perturbé et au caryotype des cellules des personnes malades et enfin l’échelle
moléculaire qui montre les protéines perturbées et ce qu’elles entraînent.
Nous verrons ensuite les différents traitements mis en place ainsi que les espoirs de ces trois
maladies.

4
A) La Progéria
Etymologiquement « progéria » vient du mot grec « geron » qui
signifie « le vieillard ». La Progéria est une maladie génétique
extrêmement rare qui touche environ, garçon comme fille, un
nouveau-né sur huit millions soit approximativement 3 cas en
France, 25 en Europe et une centaine dans le monde. Elle a été
décrite en 1886 par J. Hutchinson (à droite) et Gilford (à gauche).
Echelle macroscopique
La maladie est principalement caractérisée par un vieillissement prématuré et un
nanisme accentué. La taille ne dépasse pas 110cm pour 15Kg. La croissance des enfants
est retardée mais la peau présente un vieillissement accéléré. Ils ont une « tête
d’oiseau » (visage étroit ainsi qu’une tête disproportionnée), une hypotrichose
généralisée (diminution ou absence de poils et des cheveux). La peau devient mince,
flétrie, ridée et à certains endroits hyper pigmentée et il apparaît une cyanose
(coloration bleuâtre de la peau).
Des problèmes dentaires entraînent un micrognathisme (mâchoire de petit volume) et
les oreilles proéminentes présentent une absence de lobule. Les enfants atteints ont un
thorax en forme de poire, des anomalies articulaires et des problèmes cardiovasculaires.
Les premiers symptômes
apparaissent entre 18 et 24 mois. Dès
lors, les enfants atteints ont une
durée de vie d’environ 14,5ans.
La progéria n’affecte pas le
développement mental de l’enfant.

5
Echelle cellulaire
La progéria est la conséquence de la modification du gène lamine A sur le chromosome
1.
La mutation est présente chez les enfants
mais pas chez leurs parents. Le gène est
transmis dans la cellule œuf et en se
divisant, celle-ci a transmis ce gène à toutes
les autres cellules de l’enfant qui sont donc
atteintes et porteuses du gène morbide.
C'est une maladie récessive.
Sur le chromosome 1, en position 1824, il y a eu une
mutation de substitution d’un nucléotide : La Cytosine
présente chez un individu sain est remplacée par de la
Thymine chez un individu malade.
Echelle moléculaire
C’est une mutation de substitution. Le codon ainsi formé est « GGT » au lieu de « GGC »
mais ces deux codons donnent un même acide aminé : la Glycine.Sur le chromosome 1, le
gène LMNA code la protéine Lamine A. Dans le cas de la Progéria, cette protéine va
perdre 50 acides aminés et ainsi devenir une « Progérine ». La protéine va rester
attachée à la membrane nucléaire entraînant sa déformation.
Traitements
Trop rarissime pour lancer des traitements d'où
le terme de maladie « orpheline » car il n'existe
pas de réel traitement pour la soigner. Mais
différents tests doivent être pratiqués au moins
une fois par an et plus, tels que la mesure des
lipides et de la glycémie à jeun, de la tension
artérielle des bras et des jambes, effectuer une
visite cardiologique avec un examen physique,
faire des électrocardiogrammes... Toutefois, une
bonne alimentation, le rembourrage des
chaussures et un traitement à l'aspirine peuvent
être envisagés.
 6
7
8
9
10
11
12
6
7
8
9
10
11
12
1
/
12
100%