JTA 1999 : Prévalence et caractéristiques cliniques de la forme

Prévalence et caractéristiques cliniques de la
forme majeure de surdité de l'enfant, DFNB1, due
à une atteinte du gène de la connexine 26 :
implications pour le conseil génétique
Garabedian , F Denoyelle , S Marlini , D Weil , L Moatti et CH Petit
Introduction
La surdité neurosensorielle est un handicap extrêmement fréquent. Elle affecte environ un enfant sur mille à
la naissance et un sur mille au cours de l'enfance. Un tiers des surdités de l'enfant sont de cause
environnementale (infections et toxiques en période pré et postnatale, anoxie, prématurité), un tiers ont une
origine génétique, et un tiers sont des cas sporadiques de cause indéterminée.
Les formes héréditaires sont des maladies monogèniques : l'atteinte d'un seul gène est en cause dans
chaque forme de surdité. Cependant, de nombreux gènes sont impliqués : dans les formes de surdité
syndromiques, c'est à dire associées à d'autres pathologies ou malformations, plus de 80 gènes ont été
localisés sur les chromosomes humains (et 40 d'entre eux identifiés) (1).
Dans le domaine des surdités non syndromiques (isolées), qui représentent environ deux tiers des surdités
génétiques, beaucoup de retard a été pris dans la localisation et l'identification des gènes responsables en
raison du nombre important de gènes en cause (50 à 100 gènes attendus), de l'absence de critères cliniques
permettant de différencier entre elles des surdités dues à des gènes différents (et donc de l'absence de
phénotype caractéristique de l'atteinte de chaque gène) mais aussi des mariages fréquents entre personnes
sourdes rendant l'analyse génétique difficile.
Jusqu'en 1994, seul un gène de surdité isolée avait été localisé sur les chromosomes humains, DFNA1 (par
convention, les différents gènes de surdité isolée localisés sont appelées DFN pour les loci situés sur le
chromosome X, DFNA si la surdité est transmise sur un mode autosomique dominant, DFNB si la
transmission est autosomique récessive, et sont numérotés dans leur ordre de découverte). Depuis 1994, les
progrès dans ce domaine ont été importants avec 42 gènes localisés et 7 gènes différents identifiés, dont
celui de la forme de surdité DFNB1 : le gène de la connexine 26 (Cx26) (2). Ce gène est également
responsable d'une forme de surdité se transmettant sur un mode autosomique dominant, DFNA3, que nous
ne détaillerons pas en raison de sa rareté au sein des surdités de l'enfant (3).
Forte prévalence des mutations du gène de la connexine 26
(DFNB1)
1/4

Une surprise majeure est venue de la mise en évidence en 1997 de la large prédominance des mutations du
gène Cx26 parmi les formes de surdité congénitale. Nous avons montré dans une analyse multicentrique
menée parmi des familles provenant essentiellement de France, Nouvelle Zélande et Royaume-Uni, que la
moitié des familles atteintes d'une surdité congénitale autosomique récessive présentaient une atteinte de ce
gène (4). Des résultats identiques ont été rapportés simultanément par l'équipe de Zélante (5).
Nous avons de plus montré qu'une mutation particulière, 30delG (appelée aussi 35delG par d'autres
équipes), représentait 70% des mutations de ce gène. En 1998, Estivill et al ont confirmé la très grande
fréquence de la mutation 30delG : 2,5 à 4% de la population générale en Espagne et en Italie sont porteurs
hétérozygotes sains de cette mutation (6). La mutation 30delG est en conséquence, avec la mutation
DeltaF508 du gène CFTR (responsable de la mucoviscidose), la mutation pathogène humaine la plus
fréquente connue à ce jour.
Les études menées parmi des groupes d'enfants atteints de surdité sporadique de cause inexpliquée, ont
retrouvé une fréquence de mutations de Cx26 allant de 10% (7) à 37% (6). Toutes les études précédemment
citées ont concerné des enfants atteints de surdité congénitale sévère ou profonde. La facilité de détection
des mutations de Cx26 (gène de petite taille) donne la possibilité d'un diagnostic moléculaire accessible à un
grand nombre d'enfants sourds.
Cette possibilité est en train de bouleverser la pratique médicale quotidienne pour le diagnostic étiologique
d'une surdité isolée, et le problème du diagnostic prénatal se posera certainement dans les années à venir.
Aussi, il est maintenant important de connaître le phénotype de cette forme de surdité pour répondre aux
questions suivantes : les mutations du gène Cx26 sont-elles toujours associées à des surdités sévères ou
profondes ? à des surdités congénitales ? Deux enfants sourds présentant les même mutations, ou
appartenant à une même famille ont-ils des degrés de surdité identiques ?
Caractéristiques cliniques et radiologiques de DFNB1
Nous avons mené d'Avril 1997 à Septembre 1998 une étude prospective chez 140 enfants (provenant de 104
familles) atteints de surdité neurosensorielle autosomique récessive ou sporadique, parmi les patients de la
consultation de conseil génétique des surdités de l'H'pital Pasteur, et de l'H'pital d'Enfants
Armand-Trousseau, Paris. Les enfants étaient âgés de 4 à 20 ans (médiane 7 ans). Nous avons réalisé un
bilan clinique et paraclinique systématique afin de rechercher une surdité syndromique et de préciser le
mode de transmission (voir Tableau 1).
43/88 familles (49%) atteintes de surdité prélinguale présentaient des mutations de Cx26, versus 0/16 en cas
de surdité postlinguale (p < 0.01). Pour l'analyse phénotypique, nous avons comparé parmi les enfants
atteints de surdité prélinguale, les 54 enfants présentant des mutations sur les deux allèles de Cx26, avec 57
enfants indemnes de mutations (9 enfants porteurs d'une mutation 30delG sur un seul allèle ont été exclus
en raison du risque que certains d'entre eux, comme environ 3% de la population générale, soient porteurs
sains hétérozygotes de la mutation 30delG avec surdité d'autre cause).
Les mutations bialléliques de Cx26 étaient détectées dans tous les groupes d'enfants classés par degré de
surdité : chez 31/56 (55%) enfants présentant une surdité profonde, chez 14/29 (48%) enfants avec surdité
sévère, chez 8/19 enfants (42%) avec surdité moyenne et chez 1/7 avec surdité légère (14%). 33 enfants
étaient porteurs de la mutation 30delG à l'état homozygote (sur les deux allèles de Cx26). Ils étaient répartis
dans les 4 groupes de surdité (absence de corrélation phénotype-génotype). Les 16 autres enfants étaient
porteurs de la mutation 30delG sur un allèle et d'une autre mutation sur l'autre allèle.
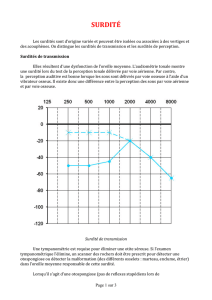
L'étude des courbes audiométriques, qui représentent la prédominance de l'atteinte fréquentielle, n'a pas
permis de retrouver de courbe pathognomonique de la forme de surdité DFNB1. Cependant la distribution
des enfants avec mutations bialléliques de Cx26 parmi les trois catégories de courbe audiométrique (Tableau
2/4

2) était significativement différente de celle des enfants indemnes de mutations (p<0.001). On peut noter
également que 2/3 des enfants ayant une courbe plate sur une oreille présentaient des mutations de Cx26,
alors que 0/11 enfants avec courbes en U ou ascendantes.
Un scanner de l'oreille interne a été pratiqué chez 23 enfants porteurs de mutations de Cx26 : aucun ne
présentait de malformation de l'oreille interne versus 7/32 enfants indemnes de mutations.
Enfin l'analyse des variations intrafamiliales a été menée chez 16 familles (35 enfants) avec mutations de
Cx26 : dans 8/16 familles, le degré de surdité était similaire au sein d'une même fratrie, alors que dans 5/16
familles, un enfant était atteint d'une surdité profonde et l'autre d'une sévère, et que dans 3/16 familles, les
enfants présentaient deux degrés de surdité d'écart (surdité moyenne chez un enfant et profonde chez
l'autre, ou légère et sévère).
Cette étude a permis de décrire le phénotype de la forme de surdité DFNB1, due à des mutations bialléliques
de Cx26 : surdité pouvant être légère à profonde, courbes audiométriques plates ou descendantes,
tomodensitométrie de l'oreille interne normale, fréquentes variations du degré de surdité à l'intérieur d'une
même famille. Ces éléments sont importants à connaître afin de proposer le diagnostic moléculaire des
mutations de Cx26 aux sujets ayant un phénotype compatible, et de donner un conseil génétique le plus
précis possible aux familles et aux sujets sourds.
Bibliographie
1. VAN CAMP G, SMITH RJH, Hereditary Hearing Loss Homepage 1998;
http://dnalab-www.uia.ac.be/dnalab/hhh.
2. KELSELL DP, DUNLOP J, STEVENS HP, et al, Ò Connexin 26 mutations in hereditary non-syndromic
sensorineural deafnes Ó. Nature, 1997 : 387, 80-3.
3. DENOYELLE F, LINA-GRANADE G, PLAUCHU H, et al, Ò Connexin26 gene linked to a dominant
deafness Ó. Nature, 1998 : 393, 319-20.
4. DENOYELLE F, WEIL D, MAW MA, et al, Ò Prelingual deafness : high prevalence of a 30delG mutation in
the connexin 26 gene Ó. Hum Mol Genet, 1997 : 6, 2173-7.
5. ZELANTE L, GASPARINI P, ESTIVILL X, et al, Ò Connexin26 mutations associated with the most
common form of non-syndromic neurosensory autosomal recessive deafness (DFNB1) in Mediterraneans Ó.
Hum Mol Genet, 1997 : 6, 1605-9.
6. ESTIVILL X, FORTINA P, SURREY S, et al, Ò Connexin-26 mutations in sporadic and inherited
sensorineural deafness Ó. Lancet, 1998 : 35, 394-8.
7. LENCH N, HOUSEMAN M, NEWTON V, et al, Ò Connexin26 mutations in sporadic non-syndromal
sensorineural deafness Ó. Lancet, 1998 : 35,
3/4

Interrogatoire dirigé : causes extrinsèqueshématuries, problèmes visuels nocturnes (rétinites dont S. de
Usher)âge de la marche (retardé dans les troubles vestibulaires)antécédents familiaux :surditéanomalies
branchiales (S. Branchio-Oto-Rénal)mèches blanches, hétérochromies iriennes (S. de Waardenburg)
pathologies rénalesgoitres (S. de Pendred)Examen pédiatriqueExamen ophtalmologique avec fond d'oeil
Recherche d'hématurie-protéinurie (bandelette)Tomodensitométrie des rochers (recherche de
malformation d'oreille interne)Pour les surdités congénitales : Electrocardiogramme (S. de Jervell et
Lange-Nielsen)En cas de goitre : T3, T4, TSH, test au perchlorate
Tableau 1. Examens cliniques et paracliniques pratiqués pour le diagnostic étiologique d'une surdité
de l'enfant.
Mutations de Cx26 Forme des courbes audiométriques (sur une oreille/sur l'autre)
Desc./ Desc.(n=65) Plate / Desc.ou Plate/Plate(n=35) Autres(n=11)
Mutations de Cx26 bialléliques 30 24
Absence de mutations 35 11 11
p < 0.001. Desc. : courbe descendante (atteinte prédominante des hautes fréquences). Plate : atteinte
similaire de toutes les fréquences. Autres : courbes audiométriques en U (atteinte prédominante des
fréquences moyennes) ou ascendantes (atteinte prédominante des fréquences graves) uni ou bilatérales.
Unité de Génétique des Déficits Sensoriels, CNRS URA 1968, Institut Pasteur, 25 rue du Dr Roux, 75724 Paris cedex 15, France. Service d'ORL
pédiatrique de Chirurgie Cervicofaciale, Hôpital d'Enfants Armand-Trousseau, 26 av. du Dr Arnold Netter 75571 Paris cedex 12, France.
4/4
1
/
4
100%