SYNDROME DU QT LONG CONGÉNITAL Des

CARDIOLOGIE
ADOLESCENCE & Médecine • Décembre 2015 • numéro 10 19
SYNDROME DU QT LONG CONGÉNITAL
Des symptômes qui apparaissent à l’adolescence
Le syndrome du QT long congénital (SQTL) se définit par un allongement de
l’intervalle QT sur l’électrocardiogramme (ECG) (Fig. 1), associé à un risque éle-
vé de troubles du rythme ventriculaire graves.
INTRODUCTION
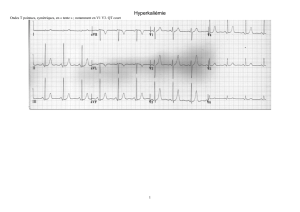
La survenue de torsades de pointes
(TDP), pouvant se dégrader en fibrilla-
tion ventriculaire, peut être à l’origine
de syncopes voire de mort subite (Fig. 2).
Dans une précédente étude, il a été mis
en évidence des mutations génétiques
impliquées dans le SQTL chez approxi-
mativement 10 % des nourrissons vic-
times de morts subites (1). Il a aussi été
démontré que plus d’un quart des décès
subits inexpliqués pourraient être en
lien avec des mutations génétiques res-
ponsables de canalopathies incluant
le SQTL (2, 3). Le SQTL est l’une des
canalopathies de transmission géné-
tique, pourvoyeuse d’arythmies poten-
tiellement létales, les plus fréquentes.
Les formes acquises se distinguent des
formes congénitales par l’apparition
d’un allongement du QTc uniquement
sous l’effet de certains médicaments ou
de perturbations hydro-électrolytiques
(hypokaliémie, hypocalcémie…).
ÉPIDÉMIOLOGIE
La prévalence du SQTL varie entre
1/2 000 et 1/5 000 individus (4, 5). Le
SQTL touche essentiellement les sujets
jeunes avec une prépondérance fémi-
nine. Une étude antérieure a démon-
tré que 50 % des patients atteints de
SQTL feront un premier évènement
cardiaque avant 12 ans (6).
PHYSIOPATHOLOGIE
ET GÉNÉTIQUE
La dépolarisation myocardique est le
résultat de la somme des dépolarisa-
tions à l’échelle cellulaire qui dépendent
en majeure partie du courant sodique
entrant dans les cellules. La repolari-
sation a pour mécanisme principal un
courant potassique sortant (Fig. 3). Les
mutations génétiques impliquées dans
le SQTL engendrent un dysfonctionne-
ment des canaux ioniques transmem-
branaires des cellules myocardiques,
qui se traduit par une perte ou un gain
de fonction de ces canaux ioniques. Le
plus souvent, il en résulte un excès de
courant sodique entrant ou un défaut de
courant potassique sortant. En consé-
quence, la durée de la dépolarisation
ou de la repolarisation ventriculaire,
Dr Linda Aïssou
CHU Avicenne, Bobigny
Figure 1 – L’intervalle QT.
Figure 2 – Torsades de pointes.
Figure 3 – Le potentiel d’action cardiaque et
ses différents courants ioniques.

CARDIOLOGIE
20 ADOLESCENCE & Médecine • Décembre 2015 • numéro 10
représentée sur l’ECG par l’intervalle
QTc, s’allonge. Cette durée varie d’une
couche myocardique à l’autre, on parle
de dispersion transmurale. Cette hété-
rogénéité de repolarisation fera le lit des
troubles du rythme ventriculaire dans
certaines conditions.
Il est recensé plusieurs sous-types de
SQTL qui se distinguent les uns des
autres par le type de gène muté. À ce
jour, plusieurs centaines de mutations
ont été décrites sur 16 gènes diffé-
rents (7). Le mode de transmission le
plus fréquent est autosomique domi-
nant (syndrome de Romano-Ward).
La forme récessive, plus rare, est l’une
des formes les plus sévères du SQTL,
elle s’associe à une surdité congénitale
neuro-sensorielle bilatérale (syndrome
de Jervell et Lange-Nielssen). Parmi les
mutations retrouvées chez les patients
atteints de SQTL, 90 % concernent les
gènes KCNQ1 (LQT1), KCNH2 (LQT2)
et SCN5A (LQT3) (8). Une mutation gé-
nétique est décelée chez 70 à 75 % des
cas de SQTL, dont le score diagnostique
de Schwartz est ≥ 4 (9). Dans approxi-
mativement 20 à 25 % des cas, aucune
mutation n’est retrouvée sur les gènes
actuellement étudiés dans le SQTL.
CRITÈRES CLINIQUES
Les symptômes se manifestent généra-
lement entre la préadolescence et la
phase adulte. La survenue de torsades
de pointes pouvant se dégrader en fi-
brillation ventriculaire peut être à l’ori-
gine de syncopes, voire de mort subite
(1 à 3 %) (10). Ces pertes de connais-
sances surviennent dans un contexte
d’effort physique, d’émotion, de stress
ou de repos. Lorsque les syncopes se
prolongent, elles peuvent s’accompa-
gner de convulsions qui peuvent être
attribuées à un diagnostic erroné de
comitialité. Dans d’autres cas, il n’est
pas rare que les patients reçoivent à
tort, un diagnostic de malaise vagal, ce
qui bien souvent entraîne un retard de
prise en charge.
Le QT corrigé par la fréquence car-
diaque (QTc) est classiquement
mesuré dans les dérivations D2 et V5
selon la formule de Bazett (QT/√RR).
Il est considéré comme allongé
lorsqu’il est supérieur à 440 msec chez
l’homme et 460 msec chez la femme.
D’après les dernières recommanda-
tions européennes, un QTc ≥ 480 msec
est retenu pour le diagnostic de SQTL
(11). L’identification du phénotype est
rendue complexe par la variabilité in-
tra-individuelle de la longueur du QTc
dans le temps. Il est donc nécessaire
de répéter les ECG chez les patients
chez qui il est suspecté un SQTL.
Dans le SQTL1, l’onde T est mono-
phasique avec une base élargie. Dans
le SQTL2, l’onde T est de faible ampli-
tude. Dans le SQTL3, l’intervalle QTc
est très allongé avec une onde T tar-
dive et de grande amplitude (Fig. 4).
La morphologie de l’onde T doit aussi
être analysée, il a été décrit un possible
crochetage de l’onde T (12), voire une
alternance des ondes T (13) (Fig. 5). Il
n’est pas rare de retrouver une brady-
cardie sinusale, en particulier chez les
enfants, ou un bloc auriculo-ventricu-
laire associé notamment chez les nou-
veau-nés atteints de SQTL.
Les circonstances de survenue d’évène-
ments cardiaques varient d’une forme à
l’autre de SQTL. Dans le SQTL1, le mode
de déclenchement est dominé par
l’effort physique (typiquement respon-
sable de noyade durant la natation) ou
le stress émotionnel (14). Dans le SQTL2,
les troubles du rythme peuvent survenir
lors d’une stimulation auditive (sonne-
rie de téléphone, bruit du réveil…), à
l’émotion, ou lors d’un réveil nocturne
(15). Dans le SQTL3, les symptômes sur-
viennent la nuit ou au repos (14).
La probabilité pré-test d’avoir un
SQTL chez un individu tout venant est
de 1/2 000, et peut aller jusqu’à 50 %
si un apparenté du premier degré a
été diagnostiqué comme génétique-
ment atteint. L’analyse de l’ECG seule
ne permet pas de poser le diagnostic
de SQTL. L’intervalle QTc dépend de
l’âge et du sexe. Il existe, par ailleurs,
une grande variabilité interindivi-
duelle, avec une dispersion de la durée
de l’intervalle QTc d’un individu à un
autre. Les courbes de distribution d’in-
tervalle QTc entre les sujets sains et les
sujets atteints se chevauchent (16).
L’évaluation d’un patient suspect de
SQTL débute par l’analyse des anté-
cédents personnels, et familiaux (avec
enquête généalogique). Il est important
de détailler les conditions précises des
pertes de connaissance, de rechercher
une mort subite inexpliquée dans la
famille. Ces éléments permettent d’ap-
précier la probabilité de SQTL devant
Figure 4 - Morphotypes de l’onde T dans le SQTL.
Figure 5 – En haut, alternance de l’onde T
chez un enfant de 2 ans porteur d’un SQTL
et ayant présenté plusieurs ACR. En bas,
encoches sur l’onde T (12, 13).

Syndrome du QT long congénital
ADOLESCENCE & Médecine • Décembre 2015 • numéro 10 21
un ECG retrouvant un allongement de
l’intervalle QT. Plus la valeur seuil du
QTc définissant le diagnostic du SQTL
est élevée et plus on perd en sensibilité
pour gagner en spécificité. Alors qu’un
QTc de 440 msec chez un parent de pre-
mier degré d’un patient atteint de SQTL
est en rapport avec un risque de SQTL de
50 %, ce risque est < à 0,1 % chez un sujet
asymptomatique sans antécédent fami-
lial. Le score de Schwartz tient compte
de ces données pour classer en probabi-
lité faible (≤ 1 point), intermédiaire (1,5 à
3 points) et forte (≥ 3,5 points) la suspi-
cion de SQTL (Tab. 1) (17).
Une fois la probabilité pré-test établie, il
convient de réaliser d’autres investiga-
tions supplémentaires comme le Holter
ECG, afin de rechercher un allongement
ou des anomalies morphologiques de
l’onde T. Le test d’effort peut parfois
déclencher des arythmies ventriculaires
(SQTL1). L’étude de l’intervalle QTc du-
rant la phase de récupération est aussi
un critère diagnostique de SQTL (18, 19).
Enfin, le gold standard reste le test géné-
tique. Cette analyse nécessite un délai
de 3 mois et n’est réalisable que dans
certains laboratoires spécialisés. Il faut
savoir que le SQTL est de pénétrance
incomplète et d’expression variable. On
peut observer des phénotypes différents
chez les membres d’une même famille
portant la même mutation.
FACTEURS DE RISQUE
Le risque d’évènements cardiaques est
plus important chez ceux qui ont sur-
vécu à un ACR, qui ont présenté une
TDP spontanée ou une syncope (20-
22). Les formes récessives sont aussi
à risque élevé. La survenue d’un pre-
mier évènement cardiaque chez les
patients de moins de 40 ans vierges de
traitement est plus importante dans le
SQTL2 ou 3. Le risque d’arythmie aug-
mente lorsque le QTc est ≥ à 500 ms, et
chez les hommes porteurs d’un SQTL3
(23). Il est décrit comme élevé chez les
femmes en postpartum, notamment
dans le SQTL2 (24).
TRAITEMENTS
Le traitement par bêtabloquant est le
traitement de première intention. Il
permet de diminuer les évènements
cardiaques de 0,97 à 0,31 par patient et
par an (25). Le nadolol est recomman-
dé à la dose de 1 mg/kg en 2 prises. Le
propranolol peut aussi être utilisé à la
dose de 2 à 4 mg/kg/j (26). L’efficacité
des bêtabloquants est clairement éta-
blie dans les SQTL 1 et 2 (14, 27, 28). Elle
reste relativement incertaine dans le
SQTL3.
La stimulation cardiaque peut-être
indiquée dans le SQTL2 en cas de bra-
dycardie ou de troubles conductifs
associés. Il est par ailleurs conseillé
de changer de sonnerie de réveil, de
téléphone… La mise en place d’un
défibrillateur automatique implan-
table est recommandée en cas de per-
sistance de syncopes ou d’arythmies
graves sous traitement bêtabloquant
maximal, ou en cas d’épisode d’ACR ré-
cupéré. Il peut être discuté chez les pa-
tients atteints du syndrome de Jervell
et Lange-Nielssen, chez les hommes
porteurs d’un SQTL3, ou lorsque le
QTc est supérieur à 500 msec.
Certains médicaments allongeant le
QT sont formellement contre-indi-
qués chez les patients atteints de SQTL
(quinidine, amiodarone, sotalol…)1.
D’autres mesures sont à appliquer,
notamment en cas de survenue de
troubles rythmiques. Il s’agit d’abord
de corriger l’hypokaliémie, et d’ap-
porter une supplémentation en ma-
gnésium. L’isuprel, voire la sonde d’en-
traînement électrosystolique externe
doivent être utilisés en cas de torsades
de pointes récidivantes.
Il est indispensable d’organiser un bi-
lan familial, puisque potentiellement
50 % des apparentés peuvent être
génétiquement atteints. Ce bilan doit
être fait dans le cadre d’une consulta-
tion spécialisée multidisciplinaire (car-
diologue, généticien, psychologue), où
seront réalisés ECG, Holter ECG et tests
génétiques. Il sera remis aux apparen-
tés porteurs de la mutation la liste des
médicaments à proscrire.
Enfin, le sport en compétition est
proscrit en dehors de la catégorie IA de
1. La liste de ces produits est consultable sur www.
qtdrugs.org ou www.cardiogen.aphp.fr.
Tableau 1 – Score de Schwartz (17).
Points
Critères ECG
QTc
≥ 480ms 3
460 –479ms 2
450 –459ms 1
QTc à la 4e minute de phase de récupération d’un test d’eort ≥ 480 ms 1
Torsades de pointes 2
Alternance de l’onde T 1
Crochetage de l’onde T dans 3 dérivations 1
Bradycardie 0,5
Antécédents cliniques
Syncope Contexte de stress 2
Sans stress 1
Surdité congénitale 0,5
Antécédents familiaux
Apparenté porteur de SQTL 1
Mort subite inexpliquée chez un apparenté de moins de 30 ans 0,5

CARDIOLOGIE
22 ADOLESCENCE & Médecine • Décembre 2015 • numéro 10
1. Arnestad M, Crotti L, Rognum TO et al. Prevalence of long-QT syndrome
gene variants in sudden infant death syndrome. Circulation2007 ; 115: 361-7.
2. Tester DJ, Medeiros-Domingo A, Will ML et al., Cardiac channel molecular
autopsy: insights from 173 consecutive cases of autopsy-negative sudden
unexplained death referred for postmortem genetic testing. Mayo Clin
Proc2012 ; 87: 524-39.
3. Tfelt-Hansen J, Winkel BG, Grunnet M, Jespersen T. Cardiac channelopathies
and sudden infant death syndrome. Cardiology2011 ; 119: 21-33.
4. Schwartz PJ, Stramba-Badiale M, Crotti L et al. Prevalence of the congenital
long-QT syndrome. Circulation2009 ; 120: 1761-7.
5. Lupoglazo JM, Denjoy I, Guicheney P. Value of genetic testing in the
management of the congenital long QT syndrome. Arch Mal Cœur Vaiss2003 ;
96: 539-47.
6. Moss AJ, Schwartz PJ, Crampton RS et al. The long QT syndrome.
Prospective longitudinal study of 328 families. Circulation1991 ; 84: 1136-44.
7. Schwartz PJ, Ackerman MJ, George AL Jr, Wilde AA. Impact of genetics on
the clinical management of channelopathies. J Am Coll Cardiol 2013 ; 62:
169-80.
8. Goldenberg I, Zareba W, Moss AJ. Long QT Syndrome. Curr Probl Cardiol
2008 ; 33: 629-94.
9. Tester DJ, Will ML, Haglund CM, Ackerman MJ. Eect of clinical phenotype on
yield of long QT syndrome genetic testing. J Am Coll Cardiol 2006 ; 47: 764-8.
10. Zareba W, Moss AJ, Schwartz PJ. Influence of genotype on the clinical
course of the long-QT syndrome. International Long-QT Syndrome Registry
Research Group. N Engl J Med 1998 ; 339: 960-5.
11. 2015 ESC Guidelines for the management of patients with ventricular
arrhythmias and the prevention of sudden cardiac death: The Task Force for
the Management of Patients with Ventricular Arrhythmias and the Prevention
of Sudden Cardiac Death of the European Society of Cardiology (ESC)
Endorsed by: Association for European Paediatric and Congenital Cardiology
(AEPC). Eur Heart J.
12. Malfatto G, Beria G, Sala S et al. Quantitative analysis of T wave
abnormalities and their prognostic implications in the idiopathic long QT
syndrome. J Am Coll Cardiol 1994 ; 23: 296-301.
13. Schwartz PJ, Malliani A. Electrical alternation of the T-wave: clinical and
experimental evidence of its relationship with the sympathetic nervous system
and with the long Q-T syndrome. Am Heart J1975 ; 89: 45-50.
14. Schwartz PJ, Priori SG, Spazzolini C et al. Genotype-phenotype correlation
in the long-QT syndrome: gene-specific triggers for life-threatening
arrhythmias. Circulation2001 ; 103: 89-95.
15. Wilde AA, Jongbloed RJ, Doevendans PA et al. Auditory stimuli as a trigger
for arrhythmic events dierentiate HERG-related (LQTS2) patients from
KVLQT1-related patients (LQTS1). J Am Coll Cardiol 1999 ; 33: 327-32.
16. Vincent GM. Role of DNA testing for diagnosis, management, and genetic
screening in long QT syndrome, hypertrophic cardiomyopathy, and Marfan
syndrome. Heart2001 ; 86: 12-4.
17. Schwartz PJ, Crotti L. QTc behavior during exercise and genetic testing for
the long-QT syndrome. Circulation2011 ; 124: 2181-4.
18. Sy RW, van der Werf C, Chattha I Set al. Derivation and validation of a
simple exercise-based algorithm for prediction of genetic testing in relatives
of LQTS probands. Circulation2011 ; 124: 2187-94.
19. Horner JM, Horner MM, Ackerman MJ. The diagnostic utility of recovery
phase QTc during treadmill exercise stress testing in the evaluation of long QT
syndrome. Heart Rhythm 2011 ; 8: 1698-704.
20. Goldenberg I, Mathew J, Moss AJ et al. Corrected QT variability in serial
electrocardiograms in long QT syndrome: the importance of the maximum
corrected QT for risk stratification. J Am Coll Cardiol 2006 ; 48: 1047-52.
21. Jons C, Moss AJ, Goldenberg I et al. Risk of fatal arrhythmic events in long
QT syndrome patients after syncope. J Am Coll Cardiol 2010 ; 55: 783-8.
22. Liu JF, Jons C, Moss AJ et al. Risk factors for recurrent syncope and
subsequent fatal or near-fatal events in children and adolescents with long QT
syndrome. J Am Coll Cardiol 2011 ; 57: 941-50.
23. Priori SG, Schwartz PJ, Napolitano C et al. Risk stratification in the long-QT
syndrome. N Engl J Med 2003 ; 348: 1866-74.
24. Seth R, Moss AJ, McNitt S et al. Long QT syndrome and pregnancy. J Am
Coll Cardiol 2007 ; 49: 1092-8.
25. Moss AJ, Zareba W, Hall WJ et al. Eectiveness and limitations of beta-
blocker therapy in congenital long-QT syndrome. Circulation2000 ; 101:
616-23.
26. Chockalingam P, Crotti L, Girardengo G et al. Not all beta-blockers
are equal in the management of long QT syndrome types 1 and 2: higher
recurrence of events under metoprolol. J Am Coll Cardiol 2012 ; 60:
2092-9.
27. Wu J, NaikiN, Ding WG et al. A molecular mechanism for adrenergic-
induced long QT syndrome. J Am Coll Cardiol 2014 ; 63: 819-27.
28. Zankov DP, Yoshida H, Tsuji K et al. Adrenergic regulation of the
rapid component of delayed rectifier K+ current: implications for
arrhythmogenesis in LQT2 patients. Heart Rhythm 2009 ; 6: 1038-46.
BIBLIOGRAPHIE
la classification des sports (Bethesda).
CONCLUSION
Le SQTL est une pathologie poten-
tiellement mortelle dont les avancées
scientifiques de ces deux dernières
décennies ont permis une meilleure
compréhension de la physiopathologie.
Le diagnostic est souvent retardé par
une labilité dans le temps de la durée de
l’intervalle QT, ou posé par excès sur des
intervalles QT « borderline ». Le test d’ef-
fort peut alors apporter des arguments
supplémentaires en faveur ou non du
diagnostic de SQTL. En cas d’anomalie
confirmée, le patient pourra être éva-
lué par un cardiologue dans un centre
de référence. Les avancées génétiques
précisant les différents mécanismes de
mutations permettent de définir les per-
sonnes les plus à risque. Actuellement,
l’arsenal thérapeutique reste limité, et
parfois lourd lorsqu’il s’agit de mise en
place d’un défibrillateur chez des sujets
jeunes. À l’avenir, les progrès génétiques
orienteront probablement des théra-
peutiques plus ciblées.
✖L’auteur déclare ne pas avoir de liens d’intérêts.
MOTS-CLÉS
Syndrome du QT long congénital, SQTL,
Intervalle QT, Bêtabloquants
1
/
4
100%