La Maladie - association IRIS

Dr I. PELLIER
Unité Immuno-Hémato-Oncologie Pédiatrique - CHU Angers
LE DEFICIT IMMUNITAIRE PRIMITIF

32
Le système immunitaire est composé
principalement de cellules* et de protéines*,
toutes nécessaires et indispensables.
Elles sont programmées pour nous défendre
contre les agressions extérieures et éliminer un
agresseur éventuel. L'agresseur, quelle que soit
sa nature (bactérie, champignon, virus ou
parasite) s'appelle "antigène*" (appelé
également « non-soi »).
Tous les constituants de l’immunité ont un
rôle défini à l’avance car génétiquement
déterminé. Ils vont interagir entre eux,
transmettre des informations sur la nature de
l’agresseur afin d’envoyer au « front » les acteurs
les mieux formés pour détruire cet antigène.
Dans un deuxième temps,
l’organisme met tout en
œuvre pour limiter cette
réponse puis l’éteindre et
se préparer déjà à la
prochaine agression. Cette
dernière donnera lieu à une réponse plus rapide
de l’organisme car déjà préparée par l’agression
antérieure.
Dans notre système immunitaire, il existe
plusieurs types de cellules de la famille des
globules blancs*(figure 1) avec des fonctions
différentes :
Les polynucléaires et les macrophages
agissent dès les premières minutes de
l’agression. Ils ont pour fonction
principale de phagocyter (« manger »)
le microbe pour le transformer et le
présenter à d’autres cellules telles que
les lymphocytes
Les lymphocytes T sans lesquels nous
ne pouvons nous défendre contre les
infections virales, fungiques
(champignons) ou parasitaires ou
certains germes de l’environnement
(figure 2).
Quels sont les composants
du système immunitaire ?
CYTOPLASME
CYTOPLASME
Granulocyte
(polynucléaire)
Monocyte
macrophage
Lymphocyte
Bou T
GRANULATION
NOYAU
NOYAU
Introduction
Antigène
Lymphocyte T
Récepteur du
lymphocyte T
OU
Lymphocytes B
Anticorps
STIMULATION LYMPHOCYTE B
SYNTHESE Ac SPECIFIQUES DE L'Ag
CAPTURE DE L’Ag ET PHAGOCYTOSE DE L'Ag
(cf figure 3)
Cellule capturant l’Ag
et le présentant à d’autres
cellules de l’immunité
Cellule de
présentation
ACTIVATION DULYMPHOCYTE T
STIMULATION LYMPHOCYTE T
dit CYTOTOXIQUE
DESTRUCTION CELLULE
DE PRESENTATION Ag
STIMULATION AUTRES
CELLULES DE L’IMMUNITE
(macrophages et lymphocytes T "helper")
INTERACTION AVEC LYMPHOCYTE T
1 2 3
Figure 2 : Action des Immunoglobulines IgG
Figure 1 : Les leucocytes, cellules du système immunitaire
(ou globules blancs)

54
Les protéines du système immunitaire sont les
immunoglobulines (ou anticorps) aidées par le complément
(figure 3). Une immunoglobuline a pour fonction de se fixer sur
l'antigène, de le neutraliser puis l'association antigène-anticorps
va former un complexe facilement repérable par les cellules de
l'immunité. Cette reconnaissance conduit finalement à
l'élimination de l’antigène.
Chez
l’
homme
il
existe
5
classes
distinctes
d’immunoglobulines (Ig)
IgG, IgA, IgM, IgE et IgD.
Les premières Ig à intervenir lors d'une agression de l'organisme
sont les IgM.
Les IgG vont opérer, quant à elles, dans un second temps.. En
effet, cette première agression sera gardée en mémoire par les
IgG (au sein des lymphocytes dits « mémoire ») pour réagir au
plus vite afin d'éviter l'infection une nouvelle fois.Ainsi lors d'une
nouvelle agression, et si le système fonctionne correctement on
ne ressent pas de signes infectieux. C'est aussi le principe des
vaccins, qui consiste à inoculer un antigène pour stimuler le
système immunitaire, ce qui permet de pouvoir réagir rapidement
face à l’antigène, sans avoir été malade au préalable. En revanche,
chaque IgG est spécifique du type d'agression. Par exemple, les
IgG anti-varicelle ne seront pas efficaces sur la maladie de la
rougeole et inversement. C'est donc une reconnaissance
spécifique qui se fait face à un antigène donné.
La
mise
en
place
de
nos
défenses
immunitaires
va
donc se
faire
en
2 phases
:
L’immunité innée : cette réponse se met en place dès les
premières heures de l’agression. Cette réponse innée est rapide,
sans mémoire et indépendante de l'antigène. Elle n’a pas besoin
de s’adapter aux agents infectieux ou aux corps étrangers.
L’immunité adaptative : cette réponse est lente, strictement
dépendante des antigènes, et permet une mémoire immunitaire.
Chaque situation différente mène à la sélection de quelques
lymphocytes qui prennent en charge le danger lorsqu’il se
présente.
Antigène
Phase 2
Complément
et lyse de la cible
Phase 1
Agglutination
par les Anticorps
(ou adhésion)
Bactérie = Antigène
Site de fixation
du complément
Site de reconnaissance
de l'anticorps
Anticorps (IgG)
Antigène
Antigène
Figure 3 : Action des Immunoglobulines IgG

76
Une des fonctions les plus importantes du
système immunitaire est de protéger l’organisme
en créant des obstacles à la dissémination d’une
infection. Ce système est (heureusement !) bien
organisé mais t complexe. Il fait donc intervenir
un nombre d’acteurs très important.
Si ce système fait défaut (quand un des
composants du système immunitaire est absent
ou défaillant), on parle de déficit immunitaire.
Les patients « immunodéficients » présentent
généralement une sensibilité accrue aux
infections.
Le déficit immunitaire
peut être provoqué par :
un défaut des cellules du système
immunitaire : le déficit immunitaire
est dit primitif (DIP)
un facteur extérieur (au système
immunitaire) susceptible d’affecter
le système immunitaire. Lorsque
le dommage est provoqué par
un facteur environnemental ou
un agent « extérieur », on parle
de déficit immunitaire secondaire
(ou acquis) ou DIS. Les causes en
sont diverses : une chimiothérapie,
la malnutrition ou des brûlures.
Il existe un grand nombre de DIP (en théorie,
quasiment autant qu’il y a “d’acteurs”,
lymphocytes T et B, polynucléaires,
complément…). Ils sont regroupés en 5 grandes
catégories, en fonction de l’atteinte qui
prédomine :
DIP humoraux* (défaut en anticorps ou
immunoglobulines), les plus fréquents
de tous les DIP,
DIP combinés (défaut des lymphocytes
et des anticorps),
Déficits de la phagocytose (défaut des
polynucléaires),
Déficit du système du complément
DIP complexes.
Qu’est ce qu’un Déficit
immunitaire primitif (DIP) ?
La Maladie
Le DIP n’est pas toujours caractérisé par une anomalie
quantitative du système immunitaire (absence d’un composant)
mais parfois pas des anomalies qualitatives de nos cellules ou
de la transmission des messages.
Les causes sont liées à l’absence (anomalie quantitative) ou
au dysfonctionnement (anomalie qualitative) d’un élément du
système de défense immunitaire, la réaction contre les
différentes agressions ne pouvant, par conséquence, se faire
correctement.
Il en résulte des infections répétées ou sévères qui peuvent
parfois mettre en jeu le pronostic vital ou endommager certains
organes tels que les poumons, les sinus, le cerveau, les oreilles,
etc.…
Les DIP sont des maladies génétiques rares (cf chapitre“Combien
de personnes en sont atteintes en France”), qui se manifestent
différemment selon le type de déficit immunitaire. Nous
connaissons la majorité des gènes responsables de ces maladies
(plus de 120 à ce jour). Certaines sont graves et peuvent s’exprimer
dès les premiers jours ou mois de vie. D’autres apparaissent
plus tardivement et s’expriment au cours de l’enfance ou de
l’adolescence. Les signes de la maladie peuvent parfois être banals
comme des otites ou des infections pulmonaires et donc ne pas
faire évoquer en premier lieu un déficit immunitaire primitif.
Plus le diagnostic de déficit est fait précocement, meilleure sera
la prise en charge thérapeutique. Les traitements administrés
auront pour but de permettre aux patients d’avoir une vie
quasiment normale pour le plus grand nombre d’entre eux.
Quelles sont les causes et les
conséquences de la maladie ?
UN PEU DE GENETIQUE POUR MIEUX COMPRENDRE…
Le rôle du système immunitaire :

98
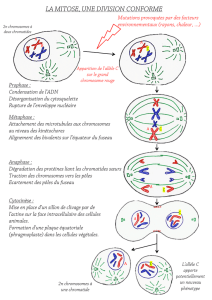
Notre organisme contient des milliards de cellules qui contiennent
une information précieuse (appelée « information génétique »)
localisée dans les gènes*(figures 4, 5). Les gènes sont contenus dans les
chromosomes* présents dans le noyau des cellules.
Chez les êtres humains,dans chaque
noyau de cellule, les chromosomes
sont au nombre de 46.
Les chromosomes allant par paires,
il en existe donc 23 paires.
22 paires sont dites “autosomes”* ou chromosomes
homologues (c’est-à-dire que les deux chromosomes sont une
copie identique pour la taille et la forme). Ils sont numérotés de
1 à 22, du plus long au plus court.
la dernière paire correspond aux deux chromosomes sexuels.
Ils sont nommés Xet Y. Ils se présentent d’une manière différente
selon le sexe de l’individu :
Chez la femme, ils sont tous les deux identiques et
appelés XX, tandis que chez l’homme, ils sont
différents : l’un est le chromosome X et l’autre est le
chromosome Y (d’où XY)(figure 6).
Qu’est ce qu’un gène
et comment les informations
sont-elles transmises ?
PROTÉINE
MEMBRANE
DE LA CELLULE
contrôlant les échanges
avec le milieu extérieur
MEMBRANE
DU NOYAU
CELLULAIRE
protégeant l’ADN
ADN
dans le noyau
CYTOPLASME
(avec des constituants
indispensables à la survie
de la cellule)
Figure 5: Cellule humaine, noyau, ADN
La forme et la taille de la cellule peuvent changer
selon le lieu où la cellule se trouve dans l’organisme.
Double hélice d’ADN
Bases azotées
(cystosine, guanine,
adénine, thymine)
Nucléotides
CHROMOSOME
Noyau
Bras long “q”
Télomère
Centromère
Bras court “p”
CELLULE
MOLECULE D’ADN
Cytoplasme
Membrane
1 tour d’hélice = 3,4 nm
2 nm
Figure 4 : De la cellule à l’ ADN. Et si on déroulait un chromosome ?
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
1
/
22
100%