Marlène Rio - MeetOchondrie

Déficits secondaires de
la chaîne respiratoire
M.Rio
Centre de Référence pour les Maladies Mitochondriales de l’enfant à l’adulte

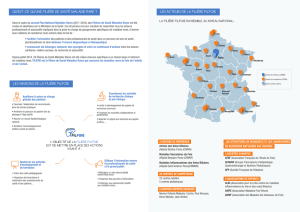
Suspecté
Sur des signes cliniques / biologiques / IRM
Basé
Sur les résultats de l’investigation enzymologique de la chaîne
respiratoire mitochondriale avec identification d’un ou des
complexes déficitaires
Confirmé
Par une étude moléculaire
Diagnostic de cytopathie mitochondriale chez l’enfant

Suspecté
Sur des signes cliniques / biologiques / IRM
Basé
Sur les résultats de l’investigation enzymologique de la chaîne
respiratoire mitochondriale avec identification d’un ou des
complexes déficitaires
Confirmé
Par une étude moléculaire
Diagnostic de cytopathie mitochondriale chez l’enfant
Peu spécifiques Inconstants Ni spécifiques ni constants
Rarement

Suspecté
Sur des signes cliniques / biologiques / IRM
Basé
Sur les résultats de l’investigation enzymologique de la chaîne
respiratoire mitochondriale avec identification d’un ou des
complexes déficitaires
Confirmé
Par une étude moléculaire
Diagnostic de cytopathie mitochondriale chez l’enfant
Peu spécifiques Inconstants Ni spécifiques ni constants
Rarement
Affirmé
Iaire ?
Secondaire?

Diagnostic de cytopathie mitochondriale chez l’enfant
Risque de porter le diagnostic de cytopathie mitochondriale
chez un enfant dont le défaut de phosphorylation oxydative
est secondaire à une autre pathologie
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
1
/
33
100%