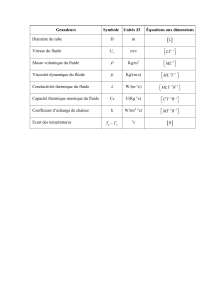
1
Modélisation thermodynamique
delamatière
E. GAGNIERE
(04.72.43.18.52)
Laboratoire d’Automatique, de Génie des
Procédés et de Génie Pharmaceutique
LAGEPP - UMR CNRS 5007 - UCB Lyon I

RAPPELSURLESBILANS
1- Principes de conservation – Bilan d’un système
« Rien ne se perd, rien ne se créé, tout se transforme » Lavoisier
Préciser le système, et sur quoi est fait le bilan
Bilans sur des quantités correspondant à un intervalle de temps donné, ou sur des
quantités par unité de temps
Exemples: bilan de matière global ou par espèce, en masse ou en mole, bilan
d’énergie, bilan d’entropie
Convention du génie des procédés :
Entrée + Production = Sortie + Consommation + Variation du contenu
>0 >0 >0 >0
Entrée/sortie =termes d’échanges avec le milieu extérieur
2 conventions pour les bilans:
Convention de la thermodynamique :
Echange + Production / Consommation = Variation du contenu
Quantités reçues ou produites : >0
Quantités cédées ou consommées : <0
2

3
Exemple de bilan:
qme (kg/s)
M(t) qms (kg/s)
Bilan de masse global (convention
GEP):
E + P = S + C + V
qme + 0 = qms + 0 + dM/dt
Système uniforme (les variables qui le
décrivent ne varient pas dans l’espace)
2- Bilan d’énergie d’un système thermodynamique uniforme
=1er principe de la thermodynamique
Energie de l’univers = cte
Création ou destruction de l’énergie = impossible
L’énergie est multiforme

4
Balle de masse M :
Energie mécanique totale
Eméca = Ec+Ep=1/2Mv2+Mgz
Diminution définitive et irréversible de
Eméca due aux forces de frottement
Force de frottement = force dissipative
ou non conservative
z
t
Ec
Ep
Ec
Ep
Eméca = 0 Eméca = 0
La balle, l’air et le sol sont de la matière qui contiennent de l’énergie
Energie interne = énergie de la matière
Disparition de Eméca correspond à une transformation en une autre forme d’énergie
Principe de conservation de l’énergie

5
Soit un système ouvert et uniforme :
système
c
1
2
kN
N-1
N-2
Termes d’échange du bilan : Entrées et sorties
: échange de chaleur à travers la paroi par unité de temps ; énergie thermique
:échange d’énergie non thermique à travers la paroi ; énergie mécanique,
électrique…(ex : )
Pour une canalisation k, on considère l’énergie transportée par la matière échangée:
c
kkkk
2
k
mk vPugz
2
υ
q
Débit
massique
kg/s
Ec Epp
Eméca (J/kg)
Énergie
interne
spécifique
du flux
Énergie mise
en jeu quand
la matière
entre ou sort
U(J) = M(kg) u(J/kg)
J/s = W
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
1
/
25
100%