Maladie de Vogt-Koyanagi-Harada : Diagnostic et Traitement
Telechargé par
Hager Bamor

21-235-B-10
Maladie
de
Vogt-Koyanagi-Harada
Vogt-Koyanagi-Harada
disease
C.
Bonnet,
J.-B.
Daudin,
A.
Brézin
Mots-clés
:
Uvéite
postérieure
Maladie
auto-immune
Décollement
séreux
rétinien
Mélanocytes
Traitement
immunosuppresseur
La
maladie
de
Vogt-Koyanagi-Harada
est
dans
sa
forme
typique
une
panuvéite
bilatérale
associée
à
des
manifestations
extraoculaires,
méningées,
cutanées
et
intéressant
l’oreille
interne.
Lors
de
la
phase
aiguë
de
la
maladie,
les
décollements
séreux
rétiniens
bilatéraux
pouvant
parfois
prendre
un
aspect
bulleux
constituent
les
éléments
les
plus
caractéristiques
conduisant
au
diagnostic.
À
la
phase
aiguë
de
la
maladie,
l’uvéite
peut
survenir
de
manière
isolée,
sans
manifestations
extraoculaires
mais
atteint
les
deux
yeux
de
fac¸on
simultanée,
argument
supplémentaire
supportant
le
diagnostic
devant
une
sémiologie
oculaire
évocatrice.
Le
pronostic
oculaire
est
bon
sous
réserve
que
le
diagnostic
soit
précoce
et
la
prise
en
charge
adaptée.
Le
traitement
standard
de
la
maladie
à
la
phase
aiguë
repose
sur
une
corticothérapie
systémique,
précoce
et
massive
à
laquelle
l’affection
est
particulièrement
sensible.
En
cas
de
retard
diagnostique
ou
de
traitement
initial
insuffisant,
des
rechutes
ou
un
passage
à
la
chronicité
sont
possibles.
Ces
formes,
responsables
de
séquelles
visuelles
parfois
importantes,
sont
de
prise
en
charge
plus
difficile,
et
requièrent
un
traitement
immunosuppresseur
dont
l’efficacité
est
inconstante.
©
2017
Elsevier
Masson
SAS.
Tous
droits
réservés.
Keywords:
Posterior
uveitis
Auto-immune
disease
Serous
retinal
detachment
Melanocytes
Treatment
by
immunosuppressants
The
Vogt-Koyanagi-Harada
disease
is
in
its
typical
form
a
bilateral
panuveitis
associated
to
extraocular,
meningeal,
cutaneous
manifestations
and
affecting
the
inner
ear.
During
the
severe
stage
of
the
disease,
the
bilateral
serous
retinal
detachments,
which
can
take
a
blistered
aspect,
constitute
the
most
characteristic
elements
leading
to
a
diagnosis.
In
the
severe
stage
of
the
disease,
the
uveitis
can
appear
in
an
isolated
manner,
without
extraocular
manifestations
but
affects
the
two
eyes
simultaneously,
providing
supplementary
argument
supporting
the
diagnosis
suggesting
an
ocular
semiology.
The
ocular
prognosis
is
good
on
condition
that
the
diagnosis
is
an
early
one
and
the
care
management
an
adapted
one.
The
standard
treatment
of
the
disease
at
a
severe
stage
relies
on
a
systemic,
early
and
massive
corticoid
therapy
to
which
the
disease
is
particularly
sensitive.
In
case
of
delayed
diagnosis
or
insufficient
initial
treatment,
relapses
or
a
change
to
chronicity
are
possible.
These
forms,
responsible
of
usually
important
visual
after-effects,
are
taken
in
charge
with
more
difficulty,
and
require
a
treatment
by
immunosuppressants
whose
efficacy
is
inconstant.
©
2017
Elsevier
Masson
SAS.
All
rights
reserved.
Plan
■Introduction
1
■Épidémiologie
et
physiopathologie
1
■Critères
diagnostiques
2
■Manifestations
oculaires
2
Segment
antérieur
2
Segment
postérieur
3
Signe
de
Sugiura
3
■Examens
complémentaires
3
Angiographies
3
Imagerie
en
tomographie
à
cohérence
optique
3
Échographie
oculaire
5
■Manifestations
extraoculaires
5
Signes
neurologiques
6
Manifestations
auditives
6
Manifestations
cutanées
7
■Diagnostics
différentiels
7
Autres
uvéites
d’origine
inflammatoire
7
Uvéites
d’origine
infectieuse
7
Sclérites
postérieures
8
■Pronostic
et
traitement
8
■Conclusion
9
Introduction
La
maladie
de
Vogt-Koyanagi-Harada
a
probablement
été
observée
dès
l’Antiquité.
Les
médecins
arabes
Ali-ibn-Isa
et
Muhammad-al-Ghâfiqî
au
Xeet
XIIesiècles
auraient
déjà
décrit
une
inflammation
oculaire
associée
à
un
vitiligo.
La
dénomina-
tion
de
la
maladie
est
liée
à
la
description
par
Vogt
en
1906
d’une
poliose
associée
à
une
inflammation
intraoculaire,
par
Harada
en
1926
de
cas
d’uvéite
postérieure
avec
décollements
de
rétine
exsu-
datifs
et
pléiocytose
à
l’analyse
du
liquide
cérébrospinal
(LCS)
et
par
Koyanagi
en
1929
de
patients
présentant
des
dépigmentations
cutanées
localisées
(vitiligo),
des
plaques
d’alopécie,
un
blanchi-
ment
des
phanères
(cheveux
et
particulièrement
cils),
associés
à
une
hypoacousie
et
à
des
acouphènes.
Dans
les
années
1930–1940,
ces
entités
avec
manifestations
d’uvéo-méningo-encéphalite
ont
été
reconnues
comme
relevant
d’un
processus
unique
et
ont
été
regroupées
sous
la
dénomination
de
syndrome
de
Vogt-Koyanagi-Harada [1].
Épidémiologie
et
physiopathologie
La
distribution
de
la
maladie
de
Vogt-Koyanagi-Harada
est
inégale
dans
le
monde,
constituant
une
cause
rare
d’uvéite
dans
les
populations
européennes,
mais
très
fréquente
en
Asie,
dans
les
populations
du
pourtour
méditerranéen
ou
chez
les
Amérindiens.
Les
études
de
populations
suggèrent
que
la
fréquence
augmen-
tée
de
la
maladie
est
associée
aux
flux
de
migration
via
le
détroit
de
Bering,
intéressant
les
amérindiens
d’Amérique
du
Nord,
cen-
trale
et
du
Sud.
La
maladie
de
Vogt-Koyanagi-Harada
représente
la
deuxième
cause
de
panuvéite
dans
la
plupart
des
séries
asia-
tiques
(15,9
%
en
Chine,
9,7
%
au
Japon)
et
la
première
cause
en
Amérique
du
Sud,
notamment
en
Argentine
(32
%
des
panu-
véites)
contre
seulement
1
à
4
%
aux
États-Unis [2–4].
Dans
une
série
de
cas
américains,
41
%
étaient
asiatiques,
21
%
blancs,
16
%
«
hispaniques
»
et
14
%
noirs.
En
France,
la
maladie
de
Vogt-
Koyanagi-Harada
représentait
environ
2
%
de
l’ensemble
des
cas
d’uvéite
vus
dans
un
centre
de
référence
et
5,9
%
des
uvéites
pos-
térieures [5].
Une
prédominance
féminine
est
notée
dans
la
plupart
des
études
et
l’âge
moyen
de
la
maladie
est
autour
de
35
ans
dans
la
plupart
des
études
(Tableau
1).
EMC
-
Ophtalmologie 1
Volume
14
>
n◦2
>
juin
2017
http://dx.doi.org/10.1016/S0246-0343(16)74282-1
© 2017 Elsevier Masson SAS. Tous droits réservés. - Document téléchargé le 02/04/2017 par SCD UNIVERSITE PAUL SABATIER TOULOUSE III (15392). Il est interdit et illégal de diffuser ce document.

21-235-B-10 Maladie
de
Vogt-Koyanagi-Harada
Tableau
1.
Principales
séries
de
patients
présentant
une
maladie
de
Vogt-Koyanagi-Harada
:
caractéristiques
générales.
Auteurs
Nombre
de
patients
Pays
Classification
selon
critères
de
2001
Sexe
(%
femmes)
Âge
moyen
de
début
(ans)
Forme
complète
Forme
incomplète
Forme
possible
Read
et
al. [6] 101
États-Unis
ND
67
35,2
Bykhovskaya
et
al. [7] 24
États-Unis
ND
79
35
Kitamura
et
al. [8] 169
Japon
11,8
%
71
%
8,9
%
56
44,7
Touitou
et
al. [5] 22
France
ND
77
33,5
Yang
et
al. [9] 410
Chine
66,6
%
18,5
%
14,9
%
47,5
35,2
Abad
et
al. [10] 11
France
9
%
81
%
9
%
45
33,5
Chee
et
al. [11] 67
Singapour
ND
59,7
42,3
Tableau
2.
Maladie
de
Vogt-Koyanagi-Harada
et
typage
human
leukocyte
antigen
(HLA)
de
classe
II.
Auteurs
Population
Principaux
résultats
Islam
et
al. [15] Japonaise
DR4
:
93
%
des
patients
(RR
:
17,4),
DRQ4
:
83
%
(RR
:
9,9)
HLA-DQAl*0301
:
100
%
(RR
:
56,5)
Weisz
et
al. [14] Hispaniques
Californie
DR4
:
56
%
des
patients
(RR
:
1,96)
DR1
:
36
%
des
patients
(RR
:
4,11)
Abad
et
al. [10] France DRB1*04
:
35
%
des
patients,
dont
DRB1*0405
:
71
%
Levinson
et
al. [13] Amérindiens
Mestizos
Californie
DRB1*04
chez
22
patients
(75,9
%)
(RR
de
l’ensemble
des
allèles
DR4
:
2,60)
Goldberg
et
al. [16] Brésilienne DR4
:
54,1
%
des
patients
(RR
:
5,116)
DRB1*0405
prédominant
(RR
:
11,76)
RR
:
risque
relatif.
La
maladie
est
présumée
liée
à
une
auto-immunité
à
média-
tion
cellulaire
dirigée
contre
les
mélanocytes
présents
dans
le
tissu
uvéal.
L’association
à
un
vitiligo
relèverait
du
même
pro-
cessus,
ainsi
que
l’atteinte
de
l’oreille
interne,
aux
sites
où
des
cellules
contenant
de
la
mélanine
sont
présentes.
Les
lympho-
cytes
CD4+(T-Helpers)
et
certaines
cytokines
(interleukines
[IL]-2,
6,
23
et
interféron
[IFN]
gamma)
semblent
jouer
un
rôle
important
dans
le
développement
de
la
maladie
par
stimulation
de
l’IL-
17,
qui
déclencherait
la
réponse
auto-immunitaire.
Le
mécanisme
jouant
un
rôle
de
«
gâchette
»
pour
déclencher
cette
inflammation
est
inconnu [12].
L’hypothèse
d’un
agent
infectieux
déclenchant
des
manifestations
auto-immunes
a
été
évoquée,
mais
n’a
pas
été
étayée.
Les
données
épidémiologiques
et
les
études
immuno-
génétiques
montrent
qu’un
terrain
génétique
est
associé
à
une
susceptibilité
augmentée
vis-à-vis
de
la
maladie,
avec
en
particu-
lier
une
liaison
à
certains
sous-types
human
leukocyte
antigen
(HLA)
de
classe
II [10,
12–14].
Les
résultats
des
principales
associations
entre
allèles
HLA
de
classe
II
et
maladie
de
Vogt-Koyanagi-Harada
sont
récapitulés
dans
le
Tableau
2[10,
13–16].
Les
mélanocytes
sont
consi-
dérés
comme
étant
des
auto-antigènes.
Ils
sont
présents
dans
les
yeux,
le
cerveau,
l’oreille
interne
et
la
peau.
Les
protéines
associées
aux
mélanocytes
tyrosine-related
protein
1
(TRP1),
TRP2,
tyrosine
et
glycoprotein
100
(gp100),
sont
les
cibles
protéiques
des
lympho-
cytes
T
auto-activés,
chez
les
patients
porteurs
du
HLA
DRB1*04
(associé
fortement
à
la
maladie).
D’autres
protéines
ne
dérivant
pas
des
mélanocytes
ont
été
mises
en
évidence
comme
cible
des
lymphocytes
T
activés [17].
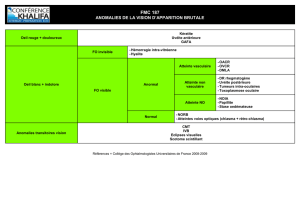
Critères
diagnostiques
La
maladie
de
Vogt-Koyanagi-Harada
a
été
l’une
des
pre-
mières
formes
d’uvéite
pour
laquelle
des
critères
diagnostiques
ont
été
proposés.
Historiquement,
les
critères
de
Sugiura
et
de
l’American
Uveitis
Society
comportaient
trois
symptômes
majeurs
pour
le
diagnostic
de
maladie
de
Vogt-Koyanagi-Harada
:
uvéite
antérieure
bilatérale,
décollements
séreux
rétiniens
observés
en
angiographie
fluorescéinique
et
pléiocytose
à
l’analyse
du
LCS.
Par
ailleurs,
les
autres
symptômes
de
la
maladie
(hypocausie,
vertiges,
poliose,
vitiligo)
étaient
considérés
comme
mineurs,
avec
une
valeur
d’appoint
pour
le
diagnostic.
Ces
trois
symp-
tômes
étaient
rarement
tous
présents,
rendant
le
diagnostic
difficile.
En
1999,
une
conférence
internationale
de
consensus
«
Vogt-
Koyanagi-Harada
Syndrome
:
First
International
Workshop
»
a
réuni
les
ophtalmologistes
experts
de
la
maladie
et
a
défini
des
critères
diagnostiques
révisés
de
la
maladie [6].
Ces
critères
per-
mettent
de
classer
les
formes
de
la
maladie
selon
leur
caractère
«
complet
»,
«
incomplet
»
ou
«
possible
»,
lesquelles
regroupent
les
atteintes
oculaires
isolées
(Tableau
3).
Dans
une
série
de
169
patients
japonais
préalablement
identifiés
en
tant
qu’atteints
de
maladie
de
Vogt-Koyanagi-Harada
selon
les
critères
de
Sugiura,
seuls
11,8
%
étaient
classés
en
tant
qu’atteints
d’une
forme
complète,
71
%
étaient
atteints
de
forme
incomplète
et
8,9
%
de
forme
probable [8].
Une
minorité
de
patients
(8,3
%),
en
par-
ticulier
ceux
ayant
subi
des
interventions
de
cataracte,
étaient
exclus
du
diagnostic
de
maladie
de
Vogt-Koyanagi-Harada.
Le
caractère
tardif
de
l’apparition
de
certaines
manifestations,
en
par-
ticulier
cutanées,
est
à
l’origine
du
classement
en
tant
que
forme
incomplète
de
nombreux
cas,
en
particulier
chez
les
patients
euro-
péens [18].
L’évolution
de
la
maladie
a
été
classiquement
décrite
en
quatre
phases
:
•
prodromale,
avec
des
manifestations
neurologiques
et
audi-
tives
;
•
uvéitique
aiguë,
avec
une
choroïdite
diffuse,
susceptible
de
se
manifester
par
des
décollements
de
rétine
exsudatifs
et
une
papillite,
avec
ou
sans
autres
signes
d’inflammation
intraocu-
laire
;
•
uvéitique
chronique,
caractérisée
par
la
survenue
variable
d’une
dépigmentation
du
fond
d’œil
(«
sunset
glow
fundus
»
ou
«
fond
d’œil
en
lueur
de
coucher
de
soleil
»)
et
du
limbe
(signe
de
Sugiura)
;
•
récidivante
chronique,
interrompant
la
phase
chronique
par
des
épisodes
d’uvéite
antérieure,
susceptible
d’être
récidivante
et/ou
chronique.
Toutefois,
d’importantes
variations
peuvent
être
observées
par
rapport
à
cette
description.
L’atteinte
est
bilatérale,
le
plus
souvent
d’installation
simultanée
des
deux
côtés
(Tableau
4).
Manifestations
oculaires
Segment
antérieur
La
présentation
la
plus
typique
de
l’uvéite
antérieure
de
la
maladie
de
Vogt-Koyanagi-Harada
est
granulomateuse,
avec
des
précipités
rétrodescémétiques
en
«
graisse
de
mouton
».
Des
synéchies
iridocristalliniennes
sont
fréquentes
et
des
nodules
de
Koeppe
ou
de
Busacca
peuvent
également
être
observés
(Fig.
1).
L’uvéite
antérieure
est
susceptible
d’avoir
une
évolution
indépen-
dante
des
manifestations
du
segment
postérieur.
Des
récidives
2EMC
-
Ophtalmologie
© 2017 Elsevier Masson SAS. Tous droits réservés. - Document téléchargé le 02/04/2017 par SCD UNIVERSITE PAUL SABATIER TOULOUSE III (15392). Il est interdit et illégal de diffuser ce document.

Maladie
de
Vogt-Koyanagi-Harada 21-235-B-10
Tableau
3.
Critères
diagnostiques
révisés
de
la
maladie
de
Vogt-Koyanagi-Harada.
1.
Absence
d’antécédent
de
traumatisme
oculaire
pénétrant
ou
de
chirurgie
oculaire
ayant
précédé
le
début
de
l’uvéite
2.
Absence
d’élément
clinique
ou
d’examen
complémentaire
évoquant
une
autre
maladie
oculaire
3.
Atteinte
oculaire
bilatérale,
pour
laquelle
les
critères
A
ou
B
doivent
être
remplis,
selon
le
stade
de
la
maladie
lorsque
le
patient
est
examiné
A.
Manifestations
précoces
de
la
maladie
(1)
Éléments
en
faveur
d’une
choroïdite
diffuse
(sans
ou
avec
une
uvéite
antérieure,
une
hyalite
ou
une
papillite)
pouvant
se
manifester
selon
l’une
des
manières
suivantes
:
a.
zones
localisées
de
liquide
sous-rétinien
b.
décollements
séreux
rétiniens
bulleux
(2)
Dans
les
cas
douteux
à
l’examen
du
fond
d’œil,
les
deux
éléments
suivants
doivent
être
également
présents
:
a.
zones
focales
de
retard
de
perfusion
choroïdienne,
zones
multifocales
de
fuite
en
tête
d’épingle,
grandes
zones
d’hyperfluorescence
en
plaque,
mélange
avec
le
liquide
sous-rétinien
et
hyperfluorescence
de
la
papille
(liste
par
ordre
d’apparition
au
cours
de
la
séquence)
en
angiographie
à
la
fluorescéine
b.
épaississement
choroïdien
diffus,
sans
signe
de
sclérite
postérieure
en
échographie
B.
Manifestations
tardives
de
la
maladie
(1)
Histoire
de
la
maladie
suggérant
un
antécédent
de
manifestations
selon
3A,
ainsi
que
soit
(2)
et
(3)
selon
ci-dessous,
ou
signes
multiples
selon
(3)
(2)
Dépigmentation
oculaire
(n’importe
laquelle
des
manifestations
ci-dessous
est
suffisante)
:
a.
fond
d’œil
en
lumière
de
coucher
de
soleil
«
sunset
glow
fundus
»
b.
signe
de
Sugiura
(3)
Autres
signes
oculaires
(n’importe
laquelle
des
manifestations
ci-dessous
est
suffisante)
:
a.
cicatrices
choroïdiennes
dépigmentées
nummulaires
b.
accumulation
et/ou
migrations
de
l’épithélium
pigmentaire
rétinien
c.
uvéite
antérieure
récidivante
ou
chronique
4.
Manifestations
neurologiques
ou
auditives
(pouvant
avoir
été
résolutives
au
moment
de
l’examen)
(n’importe
laquelle
des
manifestations
ci-dessous
est
suffisante)
:
a.
signes
méningés
(malaise,
fièvre,
céphalées,
nausées,
douleurs
abdominales,
raideur
de
la
nuque
et
du
dos
ou
combinaison
de
ces
manifestations.
Cependant,
des
céphalées
à
elles
seules
ne
peuvent
suffire
pour
définir
des
signes
méningés)
b.
acouphènes
c.
pléiocytose
à
l’examen
du
liquide
cérébrospinal
5.
Manifestations
tégumentaires
(qui
ne
peuvent
pas
avoir
précédé
le
début
de
l’atteinte
oculaire
ou
du
système
nerveux
central)
(n’importe
laquelle
des
manifestations
ci-dessous
est
suffisante)
:
a.
alopécie
b.
poliose
c.
vitiligo
Forme
complète
:
les
critères
1
à
5
doivent
être
présents.
Forme
incomplète
:
les
critères
1
à
3
doivent
être
présents
et
4
ou
5.
Maladie
possible,
forme
oculaire
isolée
:
les
critères
1
à
3
doivent
être
présents.
inflammatoires
antérieures
peuvent
être
observées,
y
compris
lorsque
toutes
les
autres
composantes
de
la
maladie
sont
contrô-
lées [19].
Segment
postérieur
À
la
phase
aiguë
de
la
maladie,
les
décollements
séreux
réti-
niens
et
l’œdème
papillaire
constituent
les
éléments
les
plus
caractéristiques
conduisant
au
diagnostic
de
maladie
de
Vogt-
Koyanagi-Harada [9,
20,
21].
L’inflammation
vitréenne
est
d’intensité
variable,
parfois
absente.
Le
décollement
séreux
rétinien
est
Tableau
4.
Intervalle
entre
l’atteinte
uni-
et
binoculaire
(d’après [9]).
Intervalle
entre
l’atteinte
uni-
et
bilatérale
Pourcentage
des
patients
Atteinte
simultanée
≈
3
jours
≈
7
jours
≈
14
jours
77,6
18,5
3,7
0,2
classiquement
multifocal,
débutant
dans
la
région
maculaire
et
pouvant
être
soit
localisé
au
pôle
postérieur
et/ou
à
la
moyenne
périphérie,
soit
étendu
jusqu’à
l’extrême
périphérie
(Fig.
2).
Il
faut
souligner
l’importance
du
décollement
séreux
rétinien
bila-
téral
survenant
dans
un
contexte
inflammatoire
comme
signe
très
évocateur
de
la
maladie.
Des
plis
rétiniens
et
choroïdiens
au
pôle
postérieur
peuvent
accompagner
l’apparition
du
décol-
lement
séreux
rétinien.
Sous
traitement,
l’évolution
est
marquée
par
une
réapplication
des
décollements
rétiniens.
Toutefois,
plu-
sieurs
complications
peuvent
influencer
le
pronostic,
comme
la
survenue
de
néovascularisation
choroïdienne
et
de
fibrose
sous-
rétinienne
(Fig.
3).
Signe
de
Sugiura
Le
signe
de
Sugiura
ou
vitiligo
périlimbique
est
décrit
chez
les
patients
asiatiques
comme
la
plus
précoce
des
manifestations
de
dépigmentation
liée
à
la
maladie.
Cette
atteinte
est
plus
rarement
ou
plus
tardivement
observée
chez
les
patients
européens.
Examens
complémentaires
Angiographies
À
la
phase
aiguë
de
la
maladie,
l’angiographie
à
la
fluores-
céine
montre
de
nombreux
points
hyperfluorescents
au
niveau
de
l’épithélium
pigmentaire.
Ces
points
augmentent
en
taille
pro-
gressivement
au
cours
de
la
séquence
angiographique
et
diffusent
autour
d’eux
dans
l’espace
sous-rétinien
et
sous
l’épithélium
pig-
mentaire.
La
fluorescéine
pénètre
ensuite
l’espace
sous-rétinien
et
délimite
les
multiples
décollements
de
rétine
exsudatifs
(Fig.
4).
Une
hyperfluorescence
papillaire
en
rapport
avec
l’œdème
papil-
laire
est
visible.
En
revanche,
les
engainements
vasculaires
rétiniens
sont
rares
et
l’imprégnation
des
parois
des
vaisseaux
ne
constitue
pas
une
caractéristique
habituelle
de
la
maladie.
À
l’angiographie
au
vert
d’indocyanine,
des
délais
de
remplis-
sage
de
la
choriocapillaire
sont
visibles,
puis
des
diffusions
à
partir
des
vaisseaux
choroïdiens
sont
observées.
À
la
phase
tardive
de
la
séquence,
les
aspects
habituels
de
la
vascularisation
choroïdienne
ne
sont
plus
visibles
et
une
hyperfluorescence
diffuse
est
observée.
Imagerie
en
tomographie
à
cohérence
optique
La
tomographie
à
cohérence
optique
(OCT)
est
actuellement
un
outil
utilisé
en
pratique
courante
pour
le
diagnostic
et
le
suivi
du
Vogt-Koyanagi-Harada.
L’imagerie
en
OCT
permet
d’observer
les
décollements
séreux
rétiniens
caractéristiques
de
la
maladie
et
leur
réapplication
après
traitement
(Fig.
5)[22–24].
Il
montre
typiquement
des
décollements
séreux
rétiniens
multifocaux
poly-
lobés
avec,
en
leur
sein,
des
cloisonnements
qui
vont
former
des
structures
membranaires.
Ces
images
permettent
de
formu-
ler
des
hypothèses
concernant
la
physiopathogénie
de
la
maladie.
Dans
un
premier
temps,
l’inflammation
choroïdienne,
cible
ini-
tiale
de
la
réaction
auto-immune,
est
responsable
d’une
rupture
de
la
barrière
hématorétinienne
interne.
Des
membranes
fibri-
neuses
se
forment
sur
l’épithélium
pigmentaire.
L’afflux
de
liquide
sous-rétinien
pousse
ces
membranes
fibrineuses
qui,
en
se
déta-
chant,
forment
des
septa,
lesquels
divisent
l’espace
sous-rétinien
en
compartiments
donnant
des
images
assez
caractéristiques [25].
Les
structures
membranaires
semblent
contenir
une
ligne
EMC
-
Ophtalmologie 3
© 2017 Elsevier Masson SAS. Tous droits réservés. - Document téléchargé le 02/04/2017 par SCD UNIVERSITE PAUL SABATIER TOULOUSE III (15392). Il est interdit et illégal de diffuser ce document.

21-235-B-10 Maladie
de
Vogt-Koyanagi-Harada
AB
Figure
1.
Synéchies
iridocristalliniennes
(A,
B)
compliquant
une
inflammation
non
contrôlée
du
segment
antérieur
au
cours
d’une
maladie
de
Vogt-Koyanagi-Harada.
ABC
DE F
G H
Figure
2.
Différents
aspects
de
décollements
séreux
rétiniens
du
pôle
postérieur,
uniques
ou
multiples
(A,
B)
plus
ou
moins
bulleux,
associés
à
un
œdème
papillaire
(C
à
F)
et
à
des
plis
cho-
roïdiens
(E,
F)
dans
le
cadre
à
la
phase
aiguë
d’une
maladie
de
Vogt-Koyanagi-Harada.
Clichés
en
mosaïque
montrant
les
décollements
séreux
rétiniens
polylobés
(G,
H).
hyperréflective
qui
est
le
prolongement
de
la
ligne
de
jonction
des
articles
internes
et
externes
des
photorécepteurs.
L’épaisseur
réti-
nienne
des
couches
internes
de
la
rétine
reste
inchangée,
quelle
que
soit
l’importance
du
décollement
séreux
rétinien [26,
27].
En
plus
de
l’atteinte
rétinienne,
qui
peut
toucher
le
pôle
pos-
térieur
ou
la
rétine
périphérique,
il
existe
très
fréquemment
un
œdème
papillaire
qui
peut
être,
lui
aussi,
analysé
en
OCT
avec
la
fonction
retinal
nerve
fiber
layer
(RNFL),
qui
met
en
4EMC
-
Ophtalmologie
© 2017 Elsevier Masson SAS. Tous droits réservés. - Document téléchargé le 02/04/2017 par SCD UNIVERSITE PAUL SABATIER TOULOUSE III (15392). Il est interdit et illégal de diffuser ce document.

Maladie
de
Vogt-Koyanagi-Harada 21-235-B-10
évidence
une
diminution
de
cet
œdème
au
cours
d’un
traitement
adapté.
L’imagerie
en
enhanced
depth
imaging-OCT
(EDI-OCT)
est
une
méthode
d’évaluation
de
la
choroïde
in
vivo,
permettant
la
mesure
de
son
épaisseur
totale
ainsi
que
ses
modifications
patho-
logiques [28].
Les
études
se
sont
intéressées
à
la
mesure
de
l’épaisseur
choroïdienne
totale,
à
la
phase
aiguë
de
la
maladie,
lors
du
traitement
initial
par
bolus
de
corticoïdes
intraveineux
et
lors
de
la
résolution
et
des
récidives
de
la
maladie [29–31].
L’inflammation
choroïdienne
est
visualisable
en
EDI-OCT
sous
la
forme
d’une
augmentation
de
l’épaisseur
choroïdienne
qui
conduit
à
une
accumulation
de
fluides
sous-rétiniens.
Hirooka
et
al. [27] ont
montré
que
la
présence
d’un
décollement
séreux
réti-
nien
à
la
phase
aiguë
de
la
maladie
est
corrélée
à
un
épaississement
choroïdien,
surtout
aux
dépens
de
la
choroïde
externe.
L’épaisseur
de
la
choroïde
diminue
après
corticothérapie
systémique,
mais
peut
réaugmenter
lors
de
récidives
des
décollements
séreux
réti-
Figure
3.
Dépigmentation,
plis
et
fibrose
sous-rétinienne
après
réappli-
cation
des
décollements
séreux
rétiniens.
niens
ou
de
l’inflammation
de
segment
antérieur [12,
23,
32].
De
même,
lors
de
passage
à
la
forme
récurrente,
l’épaisseur
cho-
roïdienne
diminue,
reflet
de
l’inflammation
chronique,
et
peut
réaugmenter
en
cas
de
récidive
inflammatoire
de
segment
anté-
rieur,
avant
les
manifestations
cliniques
antérieures [26].
La
mesure
de
l’épaisseur
choroïdienne
pourrait
être
un
marqueur
utile
pour
quantifier
l’activité
inflammatoire
de
la
maladie
et
être
prédictif
des
rechutes
inflammatoires
infracliniques.
L’analyse
OCT
en
haute
résolution
permet
d’objectiver
une
atteinte
choroïdienne,
avec
de
fines
striations
de
profil
antérieur
de
l’épithélium
pigmentaire.
La
fréquence
des
plis
choroïdiens
varie
de
12
à
65,6
%
des
cas
selon
les
séries [33].
L’augmentation
de
l’épaisseur
choroïdienne
secondaire
à
l’inflammation
entraîne
des
plis
choroïdiens
et
pousse
l’épithélium
pigmentaire
vers
la
cavité
vitréenne
car
la
rigidité
de
la
sclère
en
postérieur
ne
per-
met
pas
d’expansion
de
la
choroïde
vers
ce
versant.
Les
plis
choroïdiens,
comme
l’augmentation
de
l’épaisseur
choroïdienne,
pourraient
être
un
signe
d’inflammation
choroïdienne
sévère [34].
La
présence
de
plis
choroïdiens
est
également
associée
à
un
plus
fort
risque
de
récidive
de
la
maladie.
De
plus,
les
décollements
séreux
rétiniens
semblent
être
plus
importants
en
regard
des
plis
choroïdiens [35].
Échographie
oculaire
L’échographie
peut
être
utile
au
diagnostic
dans
des
cas
aty-
piques
et/ou
lorsque
le
fond
d’œil
est
difficilement
visible,
notamment
en
présence
de
synéchies
iridocristalliniennes
ou
d’une
cataracte.
Elle
objective
un
épaississement
choroïdien
dif-
fus
faiblement
échogène,
plus
marqué
au
niveau
de
la
région
péripapillaire
et
s’étendant
jusqu’à
la
région
équatoriale,
sans
épaississement
scléral
évident [36].
Manifestations
extraoculaires
La
fréquence
des
manifestations
extraoculaires
dans
les
principales
séries
de
patients
atteints
de
maladie
de
Vogt-
Koyanagi-Harada
est
récapitulée
dans
le
Tableau
5.
A
*
B
*
C
DEF
Figure
4.
Angiographie
à
la
fluorescéine
au
cours
de
la
maladie
de
Vogt-Koyanagi-Harada
(A
à
F)
:
diffusion
progressive
du
colorant
à
partir
des
points
hyperfluorescents,
des
pin-points
(astérisque)
(B)
précoces
jusqu’au
remplissage
de
poches
de
décollements
séreux
rétiniens
aux
temps
tardifs
(têtes
de
flèches)
(E).
Œdème
papillaire
au
temps
tardif
(tête
de
flèche)
(D).
EMC
-
Ophtalmologie 5
© 2017 Elsevier Masson SAS. Tous droits réservés. - Document téléchargé le 02/04/2017 par SCD UNIVERSITE PAUL SABATIER TOULOUSE III (15392). Il est interdit et illégal de diffuser ce document.
 6
7
8
9
10
11
6
7
8
9
10
11
1
/
11
100%