LA PHYSIOLGIE DE L’HEMOSTASE PRIMAIRE
I- Introduction :
L’hémostase est l’ensemble des mécanismes contribuant au colmatage d’une brèche vasculaire. La
blessure d’un vaisseau sanguin est un phénomène physiologique quotidien et permanant, où
l’hémorragie qui en résulte doit être arrêtée immédiatement par la formation d’un caillot temporaire
au niveau de la brèche vasculaire, afin de laisser le temps à la réparation des tissus endommagés qui
est un processus beaucoup plus lent.
L’hémostase est un phénomène instantané, très rapide, il aboutit à l’arrêt du saignement en quelques
minutes seulement après son début. C’est un processus limité au niveau de la brèche vasculaire, il est
limité également dans le temps où il s’arrête immédiatement après la formation du caillot et l’arrêt du
saignement. Toute extension de l’activation de l’hémostase en dehors de la partie endommagée du
vaisseau sanguin, ou après la formation du caillot, constitue un risque d’occlusion partielle ou totale
du vaisseau sanguin « thrombose ». En revanche toute anomalie ou déficit d’activation de l’hémostase
se traduit par un « syndrome hémorragique ».
L’hémostase se déroule en trois étapes
- L’hémostase primaire : formation d’un agrégat plaquettaire = clou plaquettaire.
- La coagulation : formation d’un caillot de fibrine solide.
- La fibrinolyse : dégradation du caillot formé.
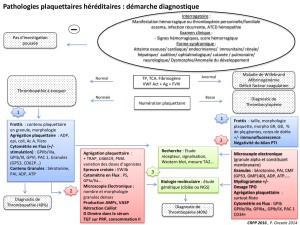
II- ETAPES DE L’HEMOSTASE PRIMAIRE :
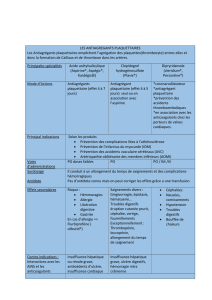
Les principaux acteurs de l’hémostase primaire sont les plaquettes sanguines ; la paroi vasculaire et un
facteur plasmatique (facteur de Von Willebrand). A l’état normal en dehors de toute blessure du
vaisseau sanguin l’hémostase primaire est inhibée ; les plaquettes restent à l’état inactif et la fluidité
du sang est maintenue sous l’effet des cellules endothéliales de la paroi vasculaire. En effet les cellules
endothéliales constituent une barrière mécanique entre le sang et le collagène sous-endothélial qui
est une substance très thrombogène. Les cellules endothéliales secrètent également des substances à
effet antiagrégant et anti-thrombotique, principalement :
*L’Oxyde d’azote NO : est un puissant agent antiagrégant plaquettaire il inhibe les plaquettes et
empêche leur activation. Les plaquettes ne tolèrent pas la présence du NO, où elles circulent dans
lumière du vaisseau sanguin loin de la concentration élevée du NO à proximité de la paroi vasculaire.
*Prostaglandine I2 : les cellules endothéliales génèrent les prostacyclines par le métabolisme des
phospholipides membranaires par le biais de leur prostacycline synthétase. La principale prostacycline
synthétisée est la prostaglandine I2 (PGI2) qui est un puissant agent antiagrégant plaquettaire et un
vasodilatateur.
*L’ADP di-phosphatase : sa sécrétion par les cellules endothéliales, dégrade les traces de l’ADP dans la
circulation, afin d’évité une activation accidentelle des plaquettes par le biais de leur récepteur
membranaire de l’ADP.
II.1- Lésion vasculaire et activation de l’hémostase primaire (temps vasculaire) :
Une lésion vasculaire déclenche une vasoconstriction réflexe de la paroi vasculaire médiée par le
système neurologique de la matrice sous endothéliale et les cellules musculaires. La vasoconstriction
locale au niveau de la brèche vasculaire minimise les pertes du sang, et diminue les forces de

cisaillement afin de permettre la formation d’un clou plaquettaire fragile au début, qui peut facilement
se détacher dans les conditions de forces de cisaillement trop importante.
La rupture de continuité des cellules endothéliales vasculaire va mettre le sang en contact direct avec
la matrice sous endothéliale ce qui activera le processus de l’hémostase primaire.
II.2- Adhésion des plaquettes au sous endothélium :
Les plaquettes arrivent en contact avec le sous endothélium vasculaire au niveau de l’endroit de la
brèche vasculaire ou elles se fixent sur le collagène sous endothéliale par l’intermédiaire du facteur de
Von Willebrand (VWF). Le VWF est un facteur plasmatique, mais également stocké au niveau des
cellules endothéliales, au moment de la lésion du vaisseau sanguin il est libéré par les cellules
endommagées, où il va se fixer d’une part au collagène et d’autre part au complexe GP Ib/IX exprimé
à la surface des plaquettes. A la fin de cette étape une couche initiale des plaquettes adhère au
collagène où cette liaison va générer un signal intracellulaire d’activation.
II.3- Activation plaquettaire :
La liaison VWF-GP Ib/IX génère un signal d’activation plaquettaire. Plusieurs phénomènes se déroulent
au cours de l’activation plaquettaire dont les principaux sont :
*Changement morphologique : les plaquettes activées perdent leur forme discoïde, où elles émettent
des pseudopodes.
*Réarrangement des phospholipides membranaires : par le phénomène de flip/flop tous les
phospholipides chargés négativement vont être exposés à la surface plaquettaire. Les plaquettes
activées constituent une surface chargée négativement indispensable pour la fixation des facteurs de
la coagulation.
*Synthèse de la Thomboxane A2 : le métabolisme des phospholipides est déclenché au moment de
l’activation plaquettaire, il aboutit à la synthèse de la thromboxane A2(TA2) par l’intermédiaire des
cyclo-oxygénase (COX) plaquettaires. La TX2 est un puissant agent agrégant plaquettaire, il active les
plaquettes et exerce un effet de chimiotactisme sur elles. Il est également un agent vasoconstricteur
qui va renforcer la vasoconstriction reflexe du vaisseau sanguin au niveau de la brèche vasculaire. Il
est à noter que les AINS et principalement l’aspirine ont un effet inhibiteur des COX plaquettaire et par
conséquence un effet antiagrégant.
*Libération du contenu granulaire : les plaquettes activées libèrent leur contenu des granules (Alpha
et denses). La sérotonine libérée à partir des granules denses renforce encore la vasoconstriction du
vaisseau, tandis que le calcium libéré est indispensable à la fixation des facteurs de la coagulation sur
la surface négative des plaquettes activées. L’ADP libéré à partir des granules denses est la substance
clé dans le processus d’agrégation plaquettaire. L’ADP agit via ses récepteurs membranaires
plaquettaires, il induit l’activation plaquettaire et l’activation du récepteur d’agrégation GPIIb/IIIa.
Il est à noter qu’à titre thérapeutique, le Clopidogrel (Plavix°) et le Ticlodipine (Ticlid°) sont utilisés pour
leur effet antiagrégant plaquettaire par le blocage des récepteurs plaquettaires de l’ADP.
II.4- Agrégation plaquettaire :
Les plaquettes activées et sensibilisées par l’ADP, expriment leur GPIIbIIIa et établissent des liaisons
entre elles par le biais du fibrinogène. Bien que le fibrinogène représente le principal liguant entre
deux GPIIbIIIIa in vivo, d’autre molécules peuvent assurer la même fonction comme : le FVW et la
fibronectine.

A la fin de cette étape, les plaquettes forment un agrégat au niveau de la brèche vasculaire, appelé :
clou plaquettaire ou bien le thrombus blanc.
Le clou plaquettaire formé est fragile, il a besoin d’une consolidation par la formation de la fibrine au
cours de la coagulation. Cependant dans les conditions de faibles forces de cisaillement au niveau de
la microcirculation, le clou plaquettaire lui seul est suffisant pour arrêter le saignement.
Comme la microcirculation est très répondue dans la partie sous cutanée et au niveau des muqueuses,
toutes anomalie de l’hémostase primaire se traduit par un syndrome hémorragique cutanéomuqueux
(Purpura).
1
/
3
100%