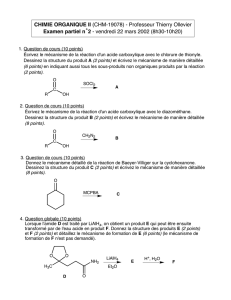
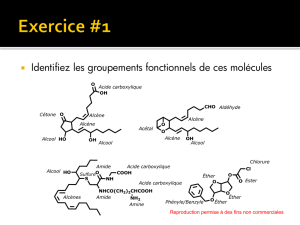
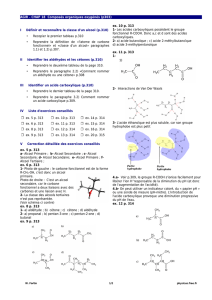
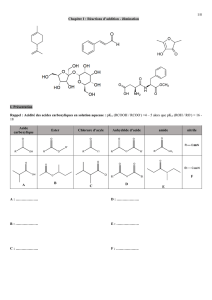
Bioisostères en chimie médicinale : Focus sur le tétrazole
Telechargé par
Anouar Alami

Page - 1 - sur 13
Intérêt des bioisostères dans la chimie médicinale
L’étude des bioisostères connaît actuellement un essor important lié à leur intérêt
considérable en tant que remplaçants de groupes fonctionnels, conduisant à des composés
biologiquement actifs. Entre les substituants hétérocycliques qui sont étudiés actuellement, le
cycle tétrazole est considéré dans la chimie médicale comme un groupe isostérique de l’acide
carboxylique.
Ce cycle contient un atome d’hydrogène acide et peut représenter une alternative
efficace de l’acide carboxylique pour concevoir des composés biologiquement actifs ayant
une résistance particulière face à la dégradation métabolique.
Ainsi, une grande partie d’aminoacides tétrazoliques présentent des propriétés
neuroexcitatrices très importantes. Des études récentes ont montré qu’ils peuvent être utilisés
dans le traitement des désordres neurologiques (épilepsie) et des maladies de
neurodégénéressence (Alzheimer et Parkinson).
Dans la littérature, peu de travaux décrivent des exemples d'α–aminoacides
tétrazoliques, on peut citer en particulier l’acide cis-4((1H-tétrazol-5-yl)méthyl) pipéridine-2-
carboxylique qui est connu pour sa grande activité antagoniste sélective du récepteur NMDA,
et la L-trans-4-(1H-tétrazol-5-yl) proline qui est un agoniste du récepteur AMPA .
La conversion d’un composé ayant une grande affinité pour une cible biologique, en
un médicament qui a du succès dans le marché représente un grand défi. Un composé qui
possède une activité pharmacologique désirable peut avoir également des caractéristiques
indésirables telles que des effets secondaires ou une toxicité.
Le bioisostérisme représente une approche utilisée par le chimiste pour essayer de
potentialiser l’activité biologique tout en essayant de réduire les effets gênants et non
souhaitables.
N
N
N
N
N
H
CO2H
H
N
N
N
N
N
H
CH2
CO2H
H
L-trans-4-(1H-tétrazol-5-yl) proline L'acide cis-4-((1H-tétrazol-5-yl) méthyl)
pipéridine-2-carboxylique

- 2 -
Un bioisostère peut être considéré comme un composé résultant de l’échange d’un
atome ou d’un groupe d’atomes par un autre atome ou un autre groupe d’atomes
généralement similaires.
Les bioisostères sont classés en bioisostères classiques et non classiques:
Les bioisostères classiques sont ceux qui possèdent les mêmes propriétés
stériques et électroniques et ont le même nombre d’atomes que le groupe pour lequel ils
sont utilisés comme remplaçants.
Les bioisostères non classiques n’obéissent pas strictement à la définition stérique
et électronique des bioisostères classiques, et n’ont pas le même nombre d’atomes
que le groupe pour lequel ils sont utilisés comme remplaçants. Ces isostères sont
capables de maintenir une activité biologique similaire à celle de la molécule grâce
au mimétisme de l’arrangement spatial du groupe fonctionnel originel ou de ses
propriétés électroniques, ou bien d’autres propriétés physico-chimiques.
I- Les bioisostères classiques
La substitution d’un atome d’hydrogène ou bien d’un groupe hydroxyle par un atome
de fluor est l’un des remplacements bioisostériques classiques les plus communément
employés. L’incorporation du fluor dans un médicament entraîne une modification des
paramètres électroniques, lipophiliques et stériques, ce qui peut influencer les propriétés
pharmacodynamiques et pharmacocinétiques du médicament.
Le rayon de van der Waals du fluor (1,47 Å) est compris entre celui de l’oxygène
(1,57 Å) et celui de l’hydrogène (1,2 Å), et le groupe trifluorométhyle a strictement la même
taille que le groupe isopropyle (2,2 Å) . Malgré le fait que le fluor est de plus grande taille que
l’hydrogène, plusieurs études ont démontré qu’il peut raisonnablement mimer l’hydrogène.
L’écart d’électronégativité est vraisemblablement la base des modifications des propriétés
pharmacologiques.
Le fluor forme une liaison solide avec le carbone (énergie de liaison = 116 Kcal/mol).
Cette liaison possède une stabilité thermique et oxydante comparée à la liaison carbone -
hydrogène (C-H = 99 Kcal/mol).
Ainsi la substitution par le fluor a été utilisée pour augmenter la période d’activité des
composés synthétiques et pour éviter la formation de métabolites toxiques.

- 3 -
De nombreux agents ayant des effets bénéfiques au niveau du système nerveux central
(SNC) contiennent un groupe CF3 ou un groupe fluorophényle. Ces groupes contribuent à
l’ensemble des activités pharmacologiques de ces composés par accroissement de leur
pénétration dans le SNC et retardent leur dégradation métabolique.
L’azétidinone (1) et son analogue fluoré (2) sont tous les deux des inhibiteurs de
l’absorption du cholestérol. Cependant l’analogue fluoré s’est montré 50 fois plus actif que
l’azétidinone (schéma 1).
Schéma 1
II- Les bioisostères non classiques
Il existe plusieurs groupes fonctionnels possédant les caractéristiques stériques et
électroniques exigées, mais ces groupes peuvent avoir des effets secondaires indésirables
comme une stabilité métabolique insuffisante ou bien une toxicité.
Les remplacements bioisostériques non classiques représentent ainsi une méthodologie
utile pour améliorer les propriétés pharmacocinétiques de la molécule cible originelle.
De nombreux bioisostères non classiques pouvant remplacer d’autres groupements
fonctionnels sont cités dans la littérature, nous décrivons brièvement les bioisostères du
groupe acide, ester, amide et phénol.
A) Les bioisostères de l’acide carboxylique
Les tétrazoles sont les bioisostères de l’acide carboxylique les plus généralement
utilisés (schéma2).
OH
O
R
O-
O
RN
N
NN
H
RN
NN
N
R
Acide carboxylique Carboxylate Tétrazole Tétrazolate
-
N
OMe
O
OMe
F
OH
F
N
O
OH
SCH 48461 (1) SCH 58235 (2)
Azétidinone

- 4 -
Schéma 2
Comme les acides carboxyliques, les tétrazoles sont ionisés au pH physiologique, et
exhibent une structure plane. les tétrazoles anioniques sont dix fois plus lipophiles que leurs
carboxylates correspondants malgré leurs acidités similaires.
Les tétrazoles 5-aryle substitués sont des acides plus forts que leurs analogues acides
benzoïques, l’anion aryle tétrazolate étant plus stable par résonance que l’anion carboxylate.
L’introduction de groupements accepteurs d’électrons à la position para du cycle 5-phényle
augmente l’acidité des acides 5-phényle tétrazoliques, et l’introduction de substituants
donneurs en position para produit un effet inverse. Ceci est en accord avec les effets des
substituants dans les acides benzoïques. Cependant, l’effet bien connu des substituants en
position ortho qui renforce l’acidité ne s’applique pas à la série des acides 5-aryle
tétrazoliques. Ceci est dû vraisemblablement à la perte de planéité et par conséquent à
l’absence de conjugaison dans les cycles.
Il faut noter que les tétrazoles 5-substitués qui contiennent une liaison N-H libre
existent sous deux formes tautomériques dans un rapport 1:1 (schéma 3).
Schéma 3
L’avantage majeur des tétrazoles sur les acides carboxyliques est qu’ils résistent à
plusieurs processus biologiques de dégradation métabolique. Les dérivés de l’acide benzoïque
subissent souvent la formation de liaisons covalentes avec les enzymes transférases pour
former des espèces activées. Le même processus d’activation ne se produit pas avec les
tétrazoles aromatiques ou aliphatiques.
Un effet pharmaceutique conservé et un profil pharmacocinétique plus favorable sont
obtenus en remplaçant le groupe carboxylate par un tétrazole métaboliquement stable.
Un bon exemple de l’utilisation du bioisostère tétrazolique est la recherche
d’antagonistes du récepteur de l’angiotensine II . la S-8307 (3) est un antagoniste sélectif du
récepteur de l’angiotensine II (schéma 4).
Schéma 4
N
NN
N
R
H
N
NN
N
R
H
(1H) (2H)
N
NCO
2
H
Cl
CO
2
H
N
NCO
2
H
Cl
Cl
EXP 6155 (4)
S - 8307 (3)

- 5 -
Les chercheurs de la société Dupont ont utilisé la modélisation moléculaire pour
ajuster la molécule (3) à la conformation active de l'angiotensine II. Ceci a conduit à la
synthèse du dérivé aryle carboxylate (4) qui a montré une augmentation significative de
l’activité sans montrer aucun effet anti-hypertensif quand il est administré par voie orale
(schéma 4).
Partant de ce point, une série de composés plus puissants a été synthétisée, comme par
exemple le composé (5) qui a montré une augmentation significative de l’affinité de liaison
pour le récepteur de l'angiotensine II et s’est montré actif par voie orale (schéma 5).
Dans l’effort de trouver des composés plus puissants et plus biodisponibles par voie
orale, une série de bioisostères de l’acide carboxylique a été préparée. A titre d'exemple, la
synthèse du biphényle tétrazole DUP 753 (Losartan) (6) (schéma 5).
Schéma 5
Losartan (6) a manifesté une meilleure affinité de liaison et une meilleure action
lorsqu’il est oralement administré. Les mêmes auteurs ont montré que l’augmentation de la
force de liaison avec le récepteur est due à la grande capacité du tétrazole à distribuer une
charge négative au pH physiologique, ce qui permet une meilleure interaction avec la charge
positive au niveau du récepteur.
- Synthèse des tétrazoles
Les méthodes classiques de synthèse des tétrazoles impliquent la réaction entre une
source d’acide hydrazoïque (azoture de sodium, chlorure d’ammonium) et un groupe
accepteur, comme le groupe nitrile, dans un solvant inerte à température élevée.
N
NOH
Cl
NNH
N
N
N
NOH
Cl
CO2H
Losartan (6)
EXP 7711 (5) DUP 753
 6
7
8
9
10
11
12
13
6
7
8
9
10
11
12
13
1
/
13
100%