Biochimie Structurale : Cours pour étudiants de 2ème année
Telechargé par
Hachani Soumaya

UNIVERSITE FERHAT ABBAS –SETIF 1
FACULTE DES SCIENCES DE LA NATURE ET DE LA VIE
DEPARTEMENT DU TRONC COMMUN
COURS DE BIOCHIMIE
STRUCTURALE
Polycopié destiné aux étudiants de 2ème année de Tronc Commun
Présenter par :
Djarmouni Meriem
2016/2017

SOMMAIRE
Introduction………………………………………………..……………...………………..…1
Chapitre I: Les liaisons chimiques…………………...…………..…..……..……….………2
1- Différents types de liaisons ………………………………………..……..…………………2
1.1. Liaisons fortes…………………………………………………...……………………..2
1.2. Liaisons faibles………………………………………….……..……………………….3
Chapitre II: les acides aminés…………………...…….………..……….…...…………....…5
II.1. Définition……………………………………..……………..……………………….……5
II.2. Classification………………………………….…………………………………………..8
II.3. Principales propriétés physiques des aminoacides……...……………………………….10
II.3.1. Stéréochimie ………………………………………………………………………......10
II.3.2. Chiralité…………………………………………………….……………………..…...11
II.3.3. Absorption dans l’ultraviolet……………………………………………………..……11
II.3.4. Ionisation………………………………………….………………………………..….12
II.3.5. Principales propriétés chimiques des acides amines...……………...…………………16
II.3.5.1. Réactions dues à la présence du groupement carboxyle……..………………...……16
II.3.5.2. Réactions dues à la présence du groupement amine……..……..……………...……17
II.3.5.2.Propriétés dues à la présence du groupement carboxyle et amine……………… .…19
Chapitre III: les peptides…………………………………..………………………......……21
III.1. Structure primaire des peptides et des protéines………...………………...................…21
III.1.1. Liaison peptidique…………………………..…………………………….…………..21
III.1.2. Détermination de la structure des chaînes peptidiques ……….…..…………….…....21
II.1.3. Fragmentation des chaînes peptidiques …………..…………………………...…....…23
III.1.4. Séparation de plusieurs chaînes peptidiques…………………......…………...………24
III.1.5. Quelques peptides ayant une importance biologique ……………………...…..……..25
Chapitre IV : les protéines……………………………..…………..…………..……….…..27
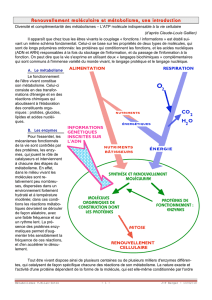
IV.1. Introduction …………………………………………………….…….…...………..…..27
IV.2. Structure des protéines…………………………………..………………………….…..28
IV.2.1. Structure primaire………………………………...…………………..………………28
IV.2.2. Structure secondaire……………………………..……………………...…….………29
IV.2.3. Structure tertiaire……………………...……………………………..…………...…..30
IV.2.4. Structure quaternaire…………………………..………………………………..…….31
IV.3. Dénaturation des protéines…………………………….…………………….……….…31
IV.4. Les techniques fondamentales utilisées pour étudier les composés protéiques………..32
IV.4.1. Chromatographie……………………………………………...………….……..……33
IV.4.1.1. Chromatographie par filtration sur gel……………….…………………..….……..33
IV.4.1.2. Chromatographie par échange d’ions…………….…………………..…………….33
IV.4.1.3. Chromatographie par interactions hydrophobes………………..……………....…..34
IV.4.1.4. Chromatographie par affinité…………………….……………………..…………..34
IV.4.2. Electrophorèse...............................................................................................................35
Chapitre V: Enzymes…………………...………………………………..……………….….36
V.1. Définition………………………….………….…………………………………..….…..36
V.2. Classification des enzymes...............................................................................................36
V.3. Site actif des enzymes…………………………..……...……………………….……….37
V.4. Notion de cinétique enzymatique…………………....…………………………………..38
V.5. Influence de différents paramètres sur la vitesse initiale………..…….……………..….40
V.5.1. Influence de la température............................................................................................40
V.5.2. Influence du pH………………………...………………………………………..…......41

LISTE DES TABLEAUX
V.6. Inhibiteurs…………………………………….…………………………..……….….....43
V.6.1. Inhibiteurs compétitifs……………………………………….…….………..………...43
V.6.2. Inhibiteurs non compétitifs............................................................................................45
V.6.3. Inhibiteurs incompétitifs…………………….……………...……….…………..….....47
V.7. L’unité enzymatique……………………………………………………………………..48
V.8. Cofacteurs……………………………………….…….…………………………..….....48
Chapitre VI: les glucides……………………………………...……………..……….…..…50
VI.1. Définition des glucides ………………………………………..……………………….50
VI.2. Importance en Biologie ……………………………….………………...………....…...50
VI.3. Classification des glucides ……………………………..………………………………50
VI.3.1. Les Oses………………………………………………………………...………....….51
VI.3.1.1. Structure linéaire des oses (Modèle de Fischer) …………………………….…..…51
VI.3.1.2. Diversité des oses, Isomérie……………… …………………………….…..……53
VI.3.1.3. Filiation des oses ……………………………………………………….………..…56
VI.3.1.4. Structure cyclique des oses……………………………………………………...….58
VI.3.1.5. propriétés physiques des oses…………………………………………….…...……60
VI.3.1.6. Propriétés chimiques des oses………………………………………………...……61
VI.4. Osides……………………………………………...……………………..……………..67
VI.4.1. Holosides…………………………………………….……………………….…..…..67
VI.4.1.1. La liaison glycosidique……………………….……………………………...…..…67
VI.4.1.2. Nomenclature et convention ………………...……………………….....……...…..67
VI.4.2. Diholosides……………………………………………………………………….…..68
VI.4.3. Polyosides……………………………………………………………………...……..69
VI.4.4. Hétérosides……………………………………….………………………..……….....72
Chapitre VII: Les lipides……………………………...…………………………...………..74
VII.1. Définition………………………..……………………………………………..……...74
VII.2. Les acides gras………………………………………………………………...…….....74
VII.2.1. Propriétés physiques des acides gras ……………………….…………..……….…..79
VII.2.2. Propriétés chimiques……………………………………………..………...………...80
VII.3. Classification des lipides………………………………….……………….…...………82
VII.3.1. Lipides simples ou ternaires………………………………………………….….......82
VII.3.1.1. Les glycérides…………………………………………...…………...……..…..….83
VII.3.1.2. Les cérides…………………………………………………………………..…..…85
VII.3.1.3. Les stérides………………………………………………………………….…….86
VII.3.2. Lipides complexes…………………………………………...……………..….…....86
VII.3.2.1. Les glycérophospholipides……………………………………………………......86
VII.3.2.2. Glycéroglycolipides…………………………………………………………..…...89
VII.3.2.3. Les sphingolipides………………………………………..………………….........89
LISTE DES FIGURES
Figure 1. Formule générale d’un acide aminé ………………………………………...………5
Figure 2 : Aminoacides à chaîne latérale non polaires………………………………..…….…9
Figure 3 : Aminoacides à chaines latérales polaires non chargées……………………...….…9
Figure 4 : Aminoacides acides…………………………………………………….…….…….10
Figure 5 : Aminoacides basiques…………………………………………………………......10
Figure 6 : Spectre d’absorption des aminoacides aromatiques dans l’ultra-violet……...…....12
Figure 7 : Courbe de titration de l’acide aminé Ala………………………………….………14
Figure 8 : Courbe de titration de Glu…………………………………...………………….…15
Figure 9 : courbe de titration de l’acide aminé basique (Lys) ……………………………....16

Figure 10 : Liaison peptidique entre deux aminoacides……………………………...…...…21
Figure 11 : Structure du glutathion réduit…………………………...……………………….25
Figure 12 : Structure de l’ocytocine et de la vasopressine…………………………………...26
Figure 13 : Structure de l’insuline………………………………………………...………....26
Figure 14 : État étiré ou structure en feuillets plissés………………………………..……....29
Figure 15 : État hélicoΪdal ou hélice α…………………………………………….………...30
Figure 16 : Structure tertiaire de la myoglobine………………………………..…………...30
Figure 17. Structure quaternaire de l’hémoglobine …………………………………...…...31
Figure 18 : Différent type de chromatographie………………………………………….….34
Figure 19 : Exemple de l’électrophorèse d’un mélange d’alanine, d’acide aspartique, et de
lysine à pH6…………………………………………………………………………………..35
Figure 20 : Variation de la vitesse initiale en fonction de la concentration en substrat, celle de
l’enzyme étant constante……………………………………………………………...……..39
Figure 21 : Représentation de Lineweaver et Burk et détermination des constantes
cinétiques…………………………………………………………………………………….40
Figure 22 : Influence de la température sur l’activité enzymatique………………………...41
Figure 23 : Influence du pH sur l’activité enzymatique………………………………….…42
Figure 24 : Influence du pH sur l’activité enzymatique de la pepsine…………………….…43
Figure 25 : Analogie structurale entre l’enzyme et l’inhibiteur compétitif……………...….43
Figure 26 : Influence d’un inhibiteur conpétitif sur l’activité enzymatique………….…..…44
Figure 27 : Fixation d’un inhibiteur non conpétitif…………………………………….........45
Figure 28 : Influence d’un inhibiteur non compétitif sur l’activité enzymatique…………...46
Figure 29 : Effets des inhibiteurs en représentation en double inverse……………………...46
Figure 30 : Influence d’un inhibiteur incompétitif sur l’activité enzymatique………………47
Figure 31 : Classification des glucides…………………………………………………...….51
Figure 32 : Filiation des D-aldoses et D-cétoses…………………………………...……..…57
Figure 33. Structure de l’acide palmitoléique………………………………...…….....……..77
Figure 34. Structure de l’acide oléique…………………………………………...…….....…77
Figure 35. Structure de l’acide linoléique……………………...……................................…78
Figure 36. Structure de l’acide alpha-linolénique………………………………...…….....…78
Figure 37. Structure de l’acide arachidonique……………………...…………………......…79
Figure 38. Disposition des micelles……………………...…..............................................…80
Figure 39. Réaction de saponification…………………………………………….....…....…80
Figure 40. Réaction d’esterification……………………...………………………….…....…80
Figure 41. Réaction d’addition d’iode…………………………………………….....…....…81
Figure 42. Oxydation des acides gras insaturés par l’acide performique………………...….81
Figure 43. Structure d’un diol……………………...……………………………………..….81
Figure 44. Oxydation puissante……………………...…....…………………………………82
Figure 45 : Structure des glycérides……………………………………………….….…..…83
Figure 46 :Structure du 1,3 distéaryl-2-palmitylglycérol ……………………….………..…84
Figure 47: Réaction de saponification……………………….………………………………85
Figure 48: Structure des cérides ……………………….………………………………....…85
Figure 49. Molécule de palmitate de cétyle. ……………………………………………..…85
Figure 50. Molécule de palmitate de cholestéryle. ……………………………..………..…86
Figure 51 :Acide phosphorique (=diacylglycérol-P)…………………………………..…….87
Figure 52 : Structure de Sphingosine………………………………………………………...89
Figure 53: Structure de Céramide…………………………………………...…………...…..90
Figure 54: Structure de sphingomyéline………………………….………...….………….....90
Figure 55 : structure de Cérébrogalactosides………………………...………....…………....91
Figure 56 : structure d’un ganglioside.…………………………………………..…………..91

Tableau 1 : Structure des acides aminés………………………………………………...…….5
Tableau 2 : Détermination de l’acide aminé en position N-terminale…………………….…22
Tableau 3 : Détermination de l’acide aminé en position C-terminale………………..……...23
Tableau 4 : Fragmentation des chaines peptidique…………………………………………..23
Tableau 5. Récapitulatif des acides gras naturels……………………………………...…….75
Tableau 6. Récapitulatif des acides gras insaturés………………………………………..….76
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
1
/
97
100%