\ Atlas de poc
cThématolo^
Diagnostic pratique
morph^ê^|to^^linique
Harald Themi
Prof. Dr. med.
Hâmatologisch-onkologische Praxisgemeinschaft und Tageskiinik
Munich
Préface de Herbert Begemann
130 planches en couleurs incluant 249 illustrations et 29 tableaux
Traduit de l'allemand par
Katharina Giinzer
Interne des hôpitaux (CHU de Caen)
Docteur Stéphane Chèze
Chef de clinique-Assistant des hôpitaux
Service d'Hématologie (CHU de Caen)
Professeur Michel Leporrier
Chef du service d'Hématologie (CHU de Caen)
Médecine-Sciences
Flammarion
4, me Casimir-Delavigne, 75006 PARIS

Dans la même collection
Atlas de poche en
couleurs
de pathologie infectieuse, par N.J. Beeching et F.J. Nye
Atlas de poche d'anatomie, par Ch. Cabrol, W. Kahie, H. Leonhardt et W. Platzer
Atlas de poche d'anatomie en coupes sériées TDM-IRM, par T.B. Môlier et E. Reif
Atlas de poche de biologie, par 1. Koolman et K.H. Rôhm
Atlas de poche de cardiologie, par A. Timmis et S. Brecker
Atlas de poche d'embryologie, par A. Drews
Atlas de poche de génétique, par E. Passarge
Atlas de poche d'histologie, par W. Kûnhel
Atlas de poche des méthodes d'analyse, par G. Schwedt
Atlas de poche des maladies sexuellement transmissibles, par A. Wisdom et D.A. Hawkins
Atlas de poche de microbiologie, par T. Hart et P. Shears
Atlas de poche de mycologie, par G. Midgiey, Y. Clayton et R.J. Hay
Atlas de poche de pharmacologie, par H. Lûllmann, K. Mohr et A. Ziegler
Atlas de poche de physiologie, par S. SilbernagI et A. Despopoulos
Atlas de poche de physiopathologie, par S. SilbernagI et F. Lang
Chez le même éditeur
Cas clinique en hématologie, par A. Najman
Aide-mémoire d'hémostase, par M. Gouault-Heilmann
Aide-mémoire de transfusion, par B. Genetet etJ.-Y. Millier
Le livre de l'interne : hématologie, par B. Varet
L'hématologie de Bernard Dreyfus, par B. Dreyfus, J. Breton-Gorius, P. Reyes
et I.-P. Vernant
Hématologie de l'enfant, par G. Schaison, A. Baruchel et T. Leblanc
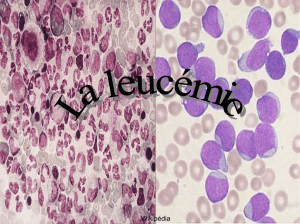
Hémopathies malignes, par R. Zittoun
L'édition originale de cet ouvrage a été publiée en allemand sous le titre :
Taschenatlas der Hamatologie. Morphologische und klinische Diagnostik fur die Praxis
© 1983, 1998, GeorgThieme Verlag, RûdigerstraRe 14, D-70469 Stuttgart.
© 2000, Flammarion Médecine-Sciences pour la traduction française.
ISBN: 2-257-10132-4

III
Sommaire
Préface, par H. Begemaim.................................................................... VII
Avant-propos........................................................................................ Vin
Avertissement pour l'édition française.............................................. X
Physiologie et physiopathologie des cellules sanguines.
Méthodes et techniques d'examen............................................... 1
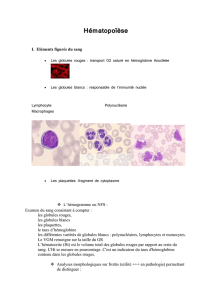
Généralités sur l'hématopoïèse normale et pathologique ............... 2
Organisation cellulaire.......................................................................... 2
Régulation et dysrégulation de l'hématopoïèse.................................... 7
Techniques d'examen et valeurs normales ....................................... 9
Prélèvement sanguin............................................................................. 9
Examen quantitatif des globules
rouges................................................
10
Détermination du taux d'hémoglobine ................................................. 11
Calcul des constantes érythrocytaires (TCMH, CCMH, VGM)........... 14
Détermination de
l'hématocrite
............................................................
14
Réticulocytes......................................................................................... 15
Numération des globules blancs
...........................................................
15
Numération des plaquettes.................................................................... 16
Valeurs normales et limites des constituants cellulaires du sang.......... 17
Frottis sanguin et son interprétation (formule sanguine)...................... 19
Intérêt de l'analyse informatisée de l'hémogramme............................. 21
Myélogramme....................................................................................... 22
Ponction ganglionnaire, ponction de tumeurs....................................... 25
Démarche diagnostique devant des anomalies sanguines ............... 27
Cellules normales du sang et des organes hématopoïétiques. 31
Cellules de l'hématopoïèse.................................................................. 32
Précurseurs de la lignée rouge : pro-érythroblastes et
érythroblastes basophiles................................................................ 32
Érythroblastes et réticulocytes.............................................................. 34
Myéloblastes et promyélocytes............................................................. 36
Myélocytes et métamyélocytes............................................................. 38
Polynucléaires neutrophiles à noyau non segmenté ou
segmenté.......................................................................................... 40
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
192
193
194
195
196
197
198
199
200
201
202
203
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
192
193
194
195
196
197
198
199
200
201
202
203
1
/
203
100%