La sclérose en plaques en 2014 Focus Multiple sclerosis in 2014

Figure 1. Les 2 événements cliniques caractérisant la sclérose
en plaques.
Poussée Progression
Images en Ophtalmologie
•
Vol. VIII
•
n
o
1
•
janvier-février 2014
10
Focus
La sclérose en plaques en 2014
Multiple sclerosis in 2014
T. Moreau, A. Fromont
(Service de neurologie, CHU de Dijon)
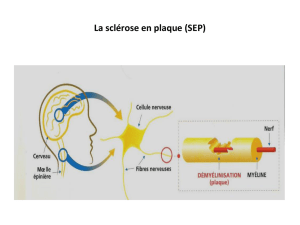
La sclérose en plaques (SEP) est une maladie
chronique diffuse du système nerveux central
(cerveau, tronc cérébral et moelle épinière). Il s’agit
d’une pathologie inflammatoire auto-immune dont
la cible est la myéline. La SEP est la première cause
de handicap neurologique du sujet jeune.
Terrain
(1-3)
La SEP affecte environ 2millions de personnes dans le monde,
et 80 000personnes en France. Elle est plus fréquente dans
l’hémisphère nord. Son incidence en France est de 7cas pour
100 000habitants et par an. Il s’agit de la maladie neurologique
la plus invalidante débutant chez l’adulte jeune, à un âge moyen
situé entre 20 et 40ans. La SEP a une prédominance féminine,
avec un sex-ratio de 3femmes pour 1homme.
Causes
Elles sont multifactorielles.
✔
Génétique
(4)
La SEP n’est pas une maladie héréditaire, mais fait intervenir
un terrain de susceptibilité génétique. Chez les jumeaux mono-
zygotes, il existe un taux de concordance maximum de 25 %.
La prévalence de la SEP dans la population générale est de
0,1 % ; elle est de 2,75 % lorsqu’un des parents est atteint et
de 4 % lorsqu’un frère ou une sœur est touché. Les formes
familiales de SEP représentent 10 % des cas. Il s’agit d’une
maladie polygénique faisant intervenir des gènes impliqués
dans la réponse immunitaire, par exemple HLA DRB1 15*01,
les gènes des récepteurs aux interleukines,etc.
✔
Environnement
(5)
Comme le prouve le modèle des migrations, l’environnement
semble intervenir dans le risque de survenue d’une SEP. Ainsi,
les personnes migrant avant l’âge de 15ans d’une zone de forte
prévalence vers une zone de faible prévalence ont un risque peu
important d’en déclarer une et inversement, ce qui n’est pas
le cas si la migration se fait à l’âge adulte. Plusieurs facteurs
environnementaux sont incriminés dans le risque de SEP :
levirus d’Epstein-Barr (mononucléose infectieuse, surtout
si elle est symptomatique), la vitamineD et l’ensoleillement,
letabac, l’obésité infantile.
Physiopathologie
(6)
La SEP est une pathologie touchant exclusivement le système
nerveux central. Des clones de lymphocytesT etB sont activés
dans l’enfance dans la circulation sanguine puis traversent,
àl’âge adulte jeune, la barrière hématoencéphalique qui isole
le système nerveux central. Une fois dans le cerveau, ces
cellules immunoactives, des cytokines pro-infl ammatoires,
desanticorps sont libérés et vont aboutir à l’attaque infl amma-
toire de la myéline. La SEP est donc une maladie infl ammatoire,
auto-immune, démyélinisante du système nerveux central.
Par l’atteinte de la myéline, le passage de l’infl ux nerveux
est touché, entraînant des signes cliniques. Les mécanismes
de remyélinisation permettent, au début, une récupération
neurologique. Une souffrance de l’axone semble coexister avec
l’atteinte myélinique.
Diagnostic
Deux événements cliniques la caractérisent : la poussée et la
progression
(fi gure1)
[7]
.
La poussée correspond à l’apparition de signes neurologiques
ou à l’aggravation de signes préexistants, durant plus de
24heures, en dehors de la fièvre, à plus de 1mois de la
dernière poussée. La progression est défi nie par l’aggravation
des signes durant 6mois et plus.
✔
Mots-clés. Diagnostic• Pronostic• Traitements.
✔
Keywords. Diagnostic• Pronostic• Treatments.

Figure 2. Évolution habituelle de la sclérose en plaques dans
le temps.
Phase rémittente Phase secondairement progressive
Figure 3. Les diff érents modes évolutifs de la sclérose en plaques.
Sans poussée surajoutée
Sclérose en plaques secondairement progressive
Avec poussées surajoutées
Sans séquelle
Sclérose en plaques rémittente
Avec séquelles
Sans poussée surajoutée
Sclérose en plaques primitivement progressive
Avec poussées surajoutées
Images en Ophtalmologie
•
Vol. VIII
•
n
o
1
•
janvier-février 2014
11
✔
Phase de début
(fi gures2 et3)
Quatre-vingt-cinq pour cent des patients démarrent leur maladie
par une forme à poussées. Ces poussées peuvent se manifester
par un ou plusieurs des signes et/ ou symptômes suivants
(8)
.
▶
Signes moteurs
Ils inaugurent la SEP dans 40 % des cas. Ils se manifestent
par une lourdeur, une faiblesse des membres. Les membres
inférieurs sont plus souvent atteints et plus précocement que
les membres supérieurs, avec souvent un défi cit asymétrique.
Lemalade n’arrive plus à courir, puis peut observer une réduc-
tion de son périmètre de marche. L’atteinte peut se limiter à des
anomalies de l’examen clinique neurologique avec un syndrome
pyramidal non défi citaire. Une trépidation épileptoïde des pieds
est souvent présente, de même qu’un signe de Babinski, qui
peut être la seule manifestation de l’atteinte pyramidale. La
spasticité des membres est fréquente ; elle aide les patients
parétiques à marcher en fauchant, mais elle est aussi à l’origine
d’une gêne et d’un inconfort.
▶
Signes sensitifs
La SEP débute par des signes sensitifs dans 45 % des cas.
Ils sont souvent subjectifs, ne correspondant pas toujours
à un dermatome. Ils peuvent être en bande autour de 1 ou
2membres, de l’abdomen ; parfois une zone de sensation
anormale en “patch” est décrite. Les signes sensitifs sont à type
d’hypoesthésie ou d’anesthésie ; il peut s’agir de signes posi-
tifs (brûlures, paresthésies, dysesthésies, engourdissements,
ruissellement, voile,etc.). Il existe un signe sensitif quasiment
spécifi que : le signe de Lhermitte (sensation de décharge élec-
trique descendant dans le dos et les membres lors de la fl exion
de la tête), témoignant d’une atteinte cordonale postérieure.
L’examinateur peut observer une atteinte de la voie lemniscale
(sensibilité épicritique et proprioceptive) et/ ou extralemniscale
(sensibilité thermoalgique et tact grossier).
Des douleurs surviennent dans la SEP (douleurs neurogènes,
dysesthésies, spasmes secondaires à la spasticité), plus
souvent dans les formes évoluées.
▶
Signes visuels
La névrite optique rétrobulbaire (NORB) est une des manifesta-
tions les plus fréquentes de la SEP lors de la première poussée
(20 % des cas). Elle se traduit par une baisse d’acuité visuelle
sur quelques heures ou quelques jours, unilatérale, à type de
voile, accompagnée de douleurs périorbitaires aggravées par les
mouvements oculaires. L’examinateur trouve une baisse d’acuité
visuelle d’importance variable avec parfois un scotome central
ou paracentral et une dyschromatopsie d’axe vert-rouge. Au
fond d’œil, la papille est le plus souvent normale au début, puis
il peut exister un fl ou du bord nasal et une hyperhémie. Un œdème
papillaire est présent dans 10 % des cas. La motricité pupillaire
intrinsèque peut être touchée. Le phénomène de Marcus Gunn
(dilatation paradoxale de la pupille du côté atteint lors de l’éclai-
rage alterné de chaque œil) témoigne d’un défi cit du réfl exe pupil-
laire afférent homolatéral ; il est fréquemment associé aux NORB.
▶
Troubles de l’équilibre, vertiges
L’atteinte vestibulaire se révèle par des sensations vertigineuses
avec signes d’instabilité d’origine vestibulaire. Les troubles
peuvent également correspondre à une atteinte cérébelleuse
(ataxie, dysmétrie, hypermétrie, adiadococinésie, asynergie,
tremblement d’intention, dysarthrie cérébelleuse, hypotonie).
▶
Extrémité céphalique
Les troubles oculomoteurs sont possibles. Un nystagmus le plus
souvent horizontal est retrouvé ; il peut être rotatoire, battant
vers le haut ou le bas. La plupart du temps, ce nystagmus est
asymptomatique, mais il peut occasionner un inconfort visuel,
une oscillopsie ou une diplopie. L’ophtalmoplégie internucléaire
(fi gure4, p.12)
uni- ou bilatérale est liée à des lésions de la
bandelette longitudinale postérieure. L’œil ipsilatéral à la lésion
ne peut pas aller en adduction, alors que l’œil controlatéral va
en abduction, mais avec un nystagmus horizontal. Les mouve-
ments de convergence sont préservés. Des anomalies de la
mobilité extrinsèque sont possibles, avec des paralysies de
latéralité ou de verticalité. La dysarthrie est fréquente. Elle est
d’origine diverse : cérébelleuse, spastique, mixte. Une névralgie
du trijumeau peut survenir au cours de la SEP. Elle peut être
révélatrice. Une paralysie faciale d’expression périphérique est
inaugurale dans 5 % des cas.

Figure 4. Ophtalmoplégie internucléaire.
Images en Ophtalmologie
•
Vol. VIII
•
n
o
1
•
janvier-février 2014
12
Focus
▶
Troubles vésicosphinctériens et sexuels
Les troubles mictionnels inaugurent la maladie chez seulement
6 % des patients, mais ils surviennent chez 78 % des patients au
cours de l’évolution de la maladie. Les patients rapportent des
pollakiuries, des mictions impérieuses, des incontinences, des
dysuries, des mictions incomplètes nécessitant des explorations
à la recherche d’un résidu postmictionnel. La constipation est
fréquente. Une incontinence fécale peut être rapportée. Les
troubles sexuels chez l’homme et chez la femme sont fréquents.
▶
Autres symptômes
La fatigue concerne environ 75 % des patients ; elle peut être inau-
gurale. Il s’agit d’un symptôme invalidant, altérant la vie quoti-
dienne
(9)
. Les troubles cognitifs concernent 40 % des patients. Ils
comprennent des troubles de l’attention, de la mémoire de travail,
du raisonnement, du maniement des concepts, de la vitesse
de traitement de l’information
(10)
. Destroubles mnésiques
affectent le stockage de l’information mais aussi le rappel différé.
Ils peuvent être précoces.
La dépression et l’anxiété touchent 27 à 54 % des patients.
▶
Symptôme durant moins de 24heures
Le phénomène d’Uhthoff correspond à une baisse transitoire, de
quelques minutes, de l’acuité visuelle déclenchée par la chaleur,
la fi èvre en période menstruelle, en postprandial. Il est présent
chez un tiers des patients et fait suite habituellement à une
NORB. Il existe des équivalents moteurs, sensitifs, oculomoteurs.
Ce phénomène traduit un bloc de conduction de l’infl ux nerveux.
Les manifestations paroxystiques consistent en des épisodes brefs
de quelques secondes, stéréotypés, déclenchés par le mouve-
ment, les stimulations sensitives, l’hyperventilation. Ces manifes-
tations peuvent être des névralgies faciales, une dysarthrie, des
mouvements anormaux paroxystiques ( dystonies, choréo- athétose
kinésigénique, akinésie paroxystique), un prurit paroxystique.
Ces différents symptômes et signes inauguraux peuvent être
isolés ou associés. Ils peuvent s’accumuler avec le temps. La
récupération après une poussée peut être complète (deux tiers
des cas endébut demaladie) ou partielle avec séquelles, qu’elle
soit traitée ou non. En moyenne, les patients ont une poussée
tous les 18mois au début, puis cette fréquence diminue.
✔
Phase d’état
(11)
En moyenne, 50 % des patients ayant une forme rémittente passe-
ront en forme secondairement progressive au bout de 10ans, avec
ou sans poussées surajoutées. Au cours du temps, il existe une
accumulation des signes et des symptômes précédents. Au-delà de
10ans d’évolution, les troubles cognitifs touchent plus de 1patient
sur 2 ; la fatigue, les troubles urinaires et sexuels sont habituels.
✔
Cas particuliers
▶
Formes progressives d’emblée(15 % des cas)
Ces formes touchent autant les hommes que les femmes.
Elles débutent à 40ans, avec un handicap qui survient plus
vite. Les formes d’emblée progressives sont habituellement
caractérisées par une atteinte médullaire progressive (réduc-
tion du périmètre de marche, troubles vésicosphinctériens).
Ces formes peuvent être avec ou sans poussées surajoutées.
▶
Formes dites “bénignes”
Certaines formes de SEP sont dites “bénignes” à moyen terme.
Il s’agit de patients encore ambulatoires après 10ans d’évo-
lution, sachant que 50 % des patients perdent leur statut de
forme bénigne dans les 10ans suivants.
Évolution et pronostic
(12)
L’évolution de la SEP va des formes dites “bénignes” aux
formes très rapidement invalidantes, avec tous les inter-
médiaires possibles.
Des courbes de survie montrent des médianes de délai
d’ atteinte des principaux niveaux de handicap évalués par
l’EDSS
(Expanded Disability Status Scale)
. Cinquante pour cent
des patients atteindront l’EDSS4 (limitation du périmètre de
marche) au bout de 11ans, l’EDSS6 (recours à une canne) après
23ans d’évolution et l’EDSS7 (fauteuil roulant) après 33ans.
Des facteurs prédictifs cliniques de bon pronostic ont été mis
en évidence, parmi lesquels un âge de début précoce, le fait
d’être une femme, un début rémittent, un début par une NORB,
un délai entre les 2premières poussées supérieur à 2ans.
Diagnostic
(13)
Le diagnostic de SEP est fondé sur un faisceau d’arguments
cliniques et paracliniques
(fi gures5 à7)
: dissémination tempo-
relle et spatiale des lésions, et atteinte du système nerveux
central non due à une autre maladie évolutive.

Figure 5. Séquences T2 fl air : coupes transverses. Hypersignaux
périventriculaires et juxtacorticaux de la substance blanche encé-
phalique.
Figure 6. Séquence T1 gadolinium : coupe coronale. Rehaussement
lésionnel annulaire par le gadolinium.
Figure 7. Séquence T1 : coupe sagittale. Atrophie globale et du corps
calleux.
Images en Ophtalmologie
•
Vol. VIII
•
n
o
1
•
janvier-février 2014
13
La dissémination temporelle des lésions se défi nit comme
la succession d’épisodes neurologiques au sein du système
nerveux central dans le temps. Elle peut être démontrée
à l’inter rogatoire, à l’examen clinique ou grâce à l’IRM par
la mise en évidence de façon contemporaine de lésions
se rehaussant par le gadolinium (signant une souffrance
du système nerveux central récente) et d’autres pas. La
survenue d’une nouvelle lésion IRM sur un nouvel examen
permet aussi d’obtenir ce critère de dissémination tempo-
relle.
La dissémination spatiale des lésions est prouvée par la mise
en évidence d’une atteinte de plusieurs zones du système
nerveux central soit à l’anamnèse ou à l’examen clinique,
soit à l’IRM. Cette quête de la dissémination spatiale et
temporelle est clinique, mais elle se fait aussi avec l’IRM.
Le diagnostic de SEP peut donc être posé devant la survenue
de 2poussées, mais, en cas de poussée unique, le critère de
dissémination temporospatiale peut être obtenu par l’IRM.
Des critères diagnostiques de la SEP dits de McDonald ont
ainsi été établis
(tableauI, p.14)
. Ces critères peuvent
permettre le diagnostic de SEP dès la première poussée
à condition que les critères de dissémination spatiale et
temporelle soient remplis.
Examens complémentaires
✔
Bilan biologique
Il n’existe aucun test biologique spécifi que pour le diagnostic
de SEP. Un bilan biologique minimal est réalisé afi n d’écarter
les diagnostics différentiels.
✔
Analyse du liquide cérébrospinal
parponctionlombaire
Elle n’est pas obligatoire pour le diagnostic, et est mainte-
nant surtout utile pour éliminer les diagnostics différentiels
de la SEP. Elle peut être normale. Dans un tiers des cas, la
cytologie révèle une pléiocytose supérieure à 5éléments
blancs par millimètre cube, mais inférieure à 50éléments,
constituée de lymphocytes ou de lymphoplasmocytes activés.
L’analyse du liquide peut permettre de démontrer le carac-
tère infl ammatoire du système nerveux central. Une hyper-
protéino rachie inférieure à 1 g/ l est retrouvée dans 25 % des
cas. Une sécrétion intrathécale d’IgG peut être démontrée
quantitativement par l’index de Link (>0,70) [rapport des
IgG du liquide céphalorachidien/sang sur rapport albumine
sang/ liquide céphalorachidien]. Cette sécrétion intrathécale
d’IgG est répartie en bandes oligoclonales mises en évidence
par immunofi xation ou isoélectrofocalisation du liquide céré-
brospinal (85 % desSEP).
✔
Potentiels évoqués
Les potentiels évoqués −visuels, sensitifs, moteurs, auditifs−
n’ont plus lieu d’être dans le diagnostic de la SEP.

Tableau I. Critères de McDonald, 2010.
Présentations cliniques Données supplémentaires afi n de poser le diagnostic de sclérose en plaques
≥ 2 poussées avec signes cliniques objectifs de 2 lésions
au moins Aucune
≥ 2 poussées avec signes cliniques objectifs d’une lésion
ET un antécédent caractéristique de sclérose en plaques
(sémiologie, évolution)
Aucune
≥ 2 poussées avec signes cliniques objectifs d’une lésion La dissémination dans l’espace pourra être retenue si :
•
l’IRM montre 1 lésion au moins dans 2 des 4 régions caractéristiques de la sclérose
en plaques (périventriculaire, juxtacorticale, sous-tentorielle*, médullaire*)
•
ou le patient présente une poussée dans un autre territoire
1 poussée avec des signes cliniques objectifs
de 2 lésions au moins La dissémination dans le temps pourra être retenue si :
•
l’IRM montre la présence simultanée de lésions asymptomatiques dont certaines
sont rehaussées par le gadolinium et d’autres non OU la présence d’une nouvelle
lésion T2 et/ou d’une nouvelle lésion prenant le gadolinium (quel que soit le délai
entre les 2 clichés)
•
ou le patient présente une nouvelle poussée
1 poussée avec des signes cliniques objectifs
d’une lésion (syndrome clinique isolé
ou premier événement démyélinisant)
La dissémination dans l’espace pourra être retenue si :
•
l’IRM montre 1 lésion au moins dans 2 des 4 régions caractéristiques de la sclérose
en plaques (périventriculaire, juxtacorticale, sous-tentorielle*, médullaire*)
•
ou le patient présente une poussée dans un autre territoire
La dissémination dans le temps pourra être retenue si :
•
l’IRM montre la présence simultanée de lésions asymptomatiques dont certaines
sont rehaussées par le gadolinium et d’autres non OU la présence d’une nouvelle
lésion T2 et/ou d’une nouvelle lésion prenant le gadolinium (quel que soit le délai
entre les 2 clichés)
•
ou le patient présente une nouvelle poussée
Aggravation progressive de symptômes neurologiques
évocateurs de sclérose en plaques (primaire progressive) Présence d’une aggravation de la maladie sur 1 an (de manière rétrospective
ou dans le cadre d’un suivi) ET 2 des 3 critères suivants :
•
mise en évidence d’une dissémination spatiale au niveau encéphalique
(≥ 1 lésion T2 dans au moins 1 région caractéristique de la sclérose en plaques
[périventriculaire, juxtacorticale, sous-tentorielle])
•
mise en évidence d’une dissémination spatiale au niveau médullaire
(≥ 2 lésions T2 médullaires)
•
mise en évidence d’une synthèse intrathécale d’immunoglobulines
(présence d’une augmentation de l’index IgG et/ou de bandes oligoclonales)
* Si le patient présente une symptomatologie médullaire ou du tronc cérébral, la/les lésion(s) symptomatique(s) n'est (ne sont) pas prise(s) en compte dans ce calcul.
Images en Ophtalmologie
•
Vol. VIII
•
n
o
1
•
janvier-février 2014
14
Focus
Diagnostics différentiels
Une atteinte au-delà du système nerveux central, c’est-à-dire
une atteinte du système nerveux périphérique, et une atteinte
systémique doivent évoquer d’autres diagnostics que celui de
la SEP.
Si certains signes sont typiques de la SEP (ophtalmoplégie
internucléaire, signe de Lhermitte, NORB), d’autres doivent être
considérés comme des “drapeaux rouges” et faire envisager
d’autres diagnostics (hémianopsie latérale homonyme, cépha-
lées, aphasie, surdité, syndrome extrapyramidal, cécité bilaté-
rale, épilepsie)
[tableauII]
. La survenue de névrites optiques
récupérant mal ou d’emblée bilatérales et/ou la survenue d’un
Tableau II. Signes typiques de la sclérose en plaques et “drapeaux
rouges” évoquant un autre diagnostic.
Signes typiques
de la sclérose en plaques
Drapeaux rouges
•
Ophtalmoplégie internucléaire
•
Signe de Lhermitte
•
Névrite optique
•
Hémianopsie latérale homonyme
•
Aphasie
•
Syndrome extrapyramidal
•
Cécité complète
•
Surdité totale
•
Atteinte du système nerveux
périphérique
•
Altération de l’état général
 6
7
6
7
1
/
7
100%