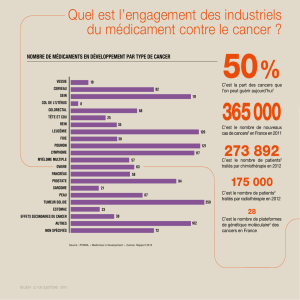
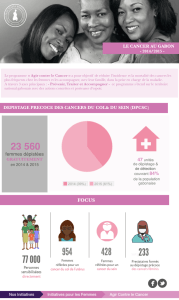
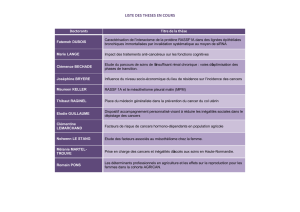
Formes familiales du cancer colorectal

Formes familiales
du cancer colorectal
Emmanuel Mitry
Fédération des spécialités digestives
CHU Ambroise Paré
AP-HP
Tlemcen, 05/05/06

épithélium!
"normal"!
mutations somatiques !
dans divers gènes !
prédispositions familiales :!
épithélium!
normal!M1 !... Mn!
M2!M3!tumeur!
mutation !
germinale!
gène-clé!
m0 / M1!
m0 / m0!
m0 / M1!m0 / m0!
mutations somatiques !
... Mn!
M2 !M3!tumeur!
cancer
sporadique : !
APPUI!
PREVENTION!


Cancérogenèse colorectale
V Boige et al. GCB 2004;28:21-32

Groupes à risque
Burt Gastroenterology 2000;119:837-53
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
1
/
35
100%