Lire l`article complet

Sujet 1 :
Amplification par PCR, puis analyse
des produits d’amplification
par électrophorèse
qui permet de distinguer les variants
du microsatellite étudié
Sujet 2 :
Sujet 3 :
Sujet : 1 2 3
(CA) x 24
(CA) x 26
(CA) x 21
Séquence microsatellite (répétition de CA)
Figure 1. Un exemple de marqueur microsatellite.
116 | La Lettre du Cancérologue • Vol. XIX - n° 2 - février 2010
DOSSIER THÉMATIQUE
Formes héréditaires
des cancers colorectaux
L
e syndrome HNPCC (Hereditary Non Polyposis
Colorectal Cancer), ou syndrome de Lynch, est
une forme de prédisposition héréditaire aux
cancers liée à la présence d’une mutation constitu-
tionnelle sur l’un des gènes du système de répara-
tion de l’ADN MMR (MisMatch Repair) [1]. Parmi le
spectre des cancers associés à ce syndrome, les deux
plus fréquents sont le cancer du côlon et le cancer de
l’endomètre. Une mutation délétère constitution-
nelle d’un des gènes MMR est identifiée chez environ
70 % des familles atteintes d’une forme typique. Le
syndrome HNPCC est responsable d’environ 3 % des
cancers colorectaux (1). Contrairement à celui de
la polypose adénomateuse familiale, le phénotype
clinique du syndrome HNPCC est beaucoup plus
hétérogène et peut mimer des situations relative-
ment fréquentes dans la population générale. Par
conséquent, ce syndrome est plus difficile à identifier
que la polypose adénomateuse familiale “classique”.
L’identification des patients atteints de ce syndrome
permet la mise en œuvre de stratégies de prévention
et de dépistage des cancers du côlon et de l’endo-
mètre afin d’améliorer la survie de ces patients.
Caractéristiques génétiques
des cancers colorectaux
associés au syndrome HNPCC
Les gènes MMR (MLH1, MSH2, MSH6 et PMS2)
codent pour des enzymes de réparation des mésap-
pariements de l’ADN (2). Le processus de carcino-
genèse observé dans le syndrome HNPCC est la
conséquence de défauts de réparation des erreurs
commises par l’ADN polymérase lors de la répli-
cation de l’ADN au cours de la division cellulaire.
Contrairement à l’inactivation d’un gène suppres-
seur de tumeur, l’inactivation d’un gène MMR n’in-
tervient pas directement dans la transformation
maligne, mais elle induit des mutations sur des gènes
cibles impliqués dans le contrôle de l’apoptose et
du cycle cellulaire. L’inactivation d’un gène MMR
confère généralement à la cellule cancéreuse un
phénotype moléculaire dit MSI+, c’est-à-dire avec
instabilité des microsatellites (3). Les microsatel-
lites sont des séquences d’ADN, généralement non
codantes, constituées de la répétition en tandem
d’un motif de 1 à 4 nucléotides (figure 1). Du fait
de leur structure répétée, les microsatellites sont
difficiles à répliquer. Au cours de la réplication de
l’ADN, ils sont des cibles privilégiées d’erreurs de
l’ADN polymérase responsables de mésappariements
de l’ADN normalement identifiés et réparés par les
enzymes du système MMR. En cas de défaillance de
ce système, on observe au niveau des microsatel-
lites une accumulation d’erreurs qui se traduit par
Le syndrome HNPCC/Lynch
HNPCC/Lynch syndrome
T. Lecomte 1, 2, G. Goujon 1, 2
1 Université François-Rabelais, Tours.
2 Service d’hépato-gastro entérologie,
hôpital Trousseau, CHRU de Tours.

Figure 2. Typage d’un marqueur microsatellite.
En cas de microsatellite polymorphe, il est nécessaire de comparer l’ADN normal à l’ADN tumo-
ral. L’analyse des produits d’amplification par électrophorèse permet de séparer les deux allèles
du microsatellite en fonction de leurs tailles (N : ADN normal, T : ADN tumoral).
Exemple de profil d’instabilité du
microsatellite monomorphe BAT26
dans le cas d’une tumeur MSI+
Microsatellite non altéré
N T
Instabilité du microsatellite
N T
La Lettre du Cancérologue • Vol. XIX - n° 2 - février 2010 | 117
Points forts
l’apparition de nouveaux allèles qui n’existent pas
à l’état constitutionnel (figure 2). Ce phénomène,
appelé MSI+, est généralement observé dans les
cellules tumorales des cancers survenant au cours
du syndrome HNPCC. Cependant, ce phénotype
moléculaire n’est pas spécifique du syndrome
HNPCC, car il est observé dans 10 à 15 % des cancers
colorectaux sporadiques, le plus souvent lié à une
hyperméthylation du promoteur du gène MLH1
conduisant à l’inactivation de ce gène (4). À noter
que l’oncogène BRAF est muté dans environ 30 %
des cancers colorectaux MSI+ sporadiques, alors
qu’il n’est pas muté dans les cancers associés au
syndrome HNPCC. De plus, les tumeurs survenant
chez les patients porteurs d’une mutation du gène
MSH6 ne présentent pas de phénotype MSI+ dans
un pourcentage de cas significatif. Classiquement,
le génotypage de cinq marqueurs microsatellites
(BAT25, BAT26, D2S123, D5S346 et D17S250)
permet la caractérisation du phénotype d’instabilité
microsatellitaire (3). Les trois marqueurs D2S123,
D5S346 et D17S250, du fait de leur caractère poly-
morphe, nécessitent le génotypage de l’ADN normal
et de l’ADN tumoral, contrairement aux marqueurs
microsatellites quasi monomorphes BAT25 et BAT26,
qui nécessitent uniquement de l’ADN tumoral pour
la recherche d’une instabilité microsatellitaire. La
recherche d’un phénotype tumoral MSI+ permet
la reconnaissance de cancers se développant dans
le cadre du syndrome HNPCC et, par conséquent,
l’identification de patients suspects d’être atteints
d’un syndrome HNPCC. Cette recherche nécessite de
l’ADN d’origine tumorale extrait à partir de matériel
tumoral (cancer colorectal ou adénome avancé)
analysable en biologie moléculaire, c’est-à-dire non
fixé dans le liquide de bouin.
Définition et présentation
clinique
Le syndrome HNPCC est une maladie à transmis-
sion autosomique dominante avec une pénétrance
comprise entre 70 et 80 %. La définition clinique du
syndrome HNPCC correspond aux critères d’Ams-
terdam établis en 1991 (Amsterdam I) puis révisés
en 1999 (Amsterdam II) [tableau I] (5). Initialement,
ces critères n’incluaient que le cancer colorectal, puis
d’autres cancers appartenant au spectre “étroit” du
syndrome HNPCC ont été intégrés dans la version
révisée (adénocarcinome de l’endomètre et de l’in-
testin grêle, carcinome urothélial). Le spectre “large”
du syndrome inclut par ailleurs les cancers de l’es-
tomac, des voies biliaires et de l’ovaire, et le glioblas-
tome. Les risques cumulés des principaux cancers
appartenant au spectre du syndrome HNPCC sont
»Le syndrome HNPCC est lié à des mutations délétères constitutionnelles des gènes MMR de réparation
de l’ADN, principalement hMSH2 et hMLH1, qui confèrent généralement à la tumeur un phénotype MSI.
»
La définition clinique du syndrome HNPCC selon les critères d’Amsterdam est peu sensible et trop
restrictive, d’où un élargissement de ces critères cliniques associé à une démarche diagnostique à deux
étapes dont la première étape est la recherche d’un phénotype tumoral MSI+ et/ou la recherche en immuno-
histochimie de la perte d’expression d’une protéine codée par les gènes MMR.
»
En cas de diagnostic de syndrome HNPCC, des mesures de prévention et de dépistage du cancer du
côlon et de l’endomètre sont à mettre en œuvre chez tous les apparentés d’une personne atteinte quand
on ne dispose pas d’un diagnostic génétique, et seulement aux porteurs d’une mutation délétère consti-
tutionnelle quand on dispose du diagnostic génétique.
Mots-clés
Syndrome HNPCC
Gènes MMR
Cancer du côlon
Chromoendoscopie
Keywords
HNPCC syndrome
MMR genes
Colorectal cancer
Chromoendoscopy
Tableau I. Critères d’Amsterdam établis par l’International Collaborative Group – HNPCC.
Critères d’Amsterdam I (critères classiques)
Famille comportant au moins trois parents atteints de cancer colorectal histologiquement prouvé et
présentant tous les critères suivants :
• l’un des sujets atteints est apparenté au premier degré aux deux autres ;
• au moins deux générations successives sont atteintes ;
• au moins un des diagnostics de cancer colorectal est posé avant l’âge de 50 ans ;
• une polypose adénomateuse familiale doit être exclue.
Critères d’Amsterdam II (critères révisés)
Famille comportant au moins trois parents atteints d’un cancer histologiquement prouvé apparte-
nant au spectre du syndrome HNPCC (cancer colorectal, cancer de l’endomètre, cancer de l’intestin
grêle, cancer de l’uretère ou des cavités rénales excrétrices) et présentant tous les critères suivants :
• l’un des sujets atteints est apparenté au premier degré aux deux autres ;
• au moins deux générations successives sont atteintes ;
• au moins un des diagnostics de cancer est posé avant l’âge de 50 ans ;
• une polypose adénomateuse familiale doit être exclue.

118 | La Lettre du Cancérologue • Vol. XIX - n° 2 - février 2010
DOSSIER THÉMATIQUE
Formes héréditaires
des cancers colorectaux Le syndrome HNPCC/Lynch
rapportés dans le tableau II (6-8). Chez les patients
atteints d’un syndrome HNPCC, le risque cumulé
de cancer, toute localisation confondue, à l’âge de
50 ans, est d’environ 60 % (7). Le risque cumulé de
cancer colorectal à 70 ans est d’environ 50 % avec un
âge moyen de survenue de 44 ans. Dans 70 % des cas,
les cancers sont localisés entre le cæcum et l’angle
colique gauche. Sur le plan anatomopathologique,
ils présentent souvent une faible différenciation, une
composante mucineuse et un infiltrat lymphoïde
important. Contrairement à ce que son nom suggère,
le syndrome HNPCC est associé à un processus de
carcinogenèse colorectale via la séquence adénome-
cancer. Le risque très élevé de cancer colorectal est
dû à une carcinogenèse colorectale accélérée à partir
du stade de l’adénome. La répartition des adénomes
sur le cadre colique est différente de celle observée
dans la population générale, avec une prédominance
entre le cæcum et l'angle colique gauche. Comme
dans la population générale, les adénomes plans
sont fréquents et, en raison de leur caractère plus
agressif, ils ont probablement un rôle important
dans le processus de carcinogenèse colorectale lié
au syndrome HNPCC. Il est à noter que le phénotype
MSI+ est observé dès le stade d’adénome avancé au
cours du syndrome HNPCC. Le risque cumulé de déve-
lopper un cancer colorectal métachrone, 20 ans après
un premier cancer colorectal, est d’environ 50 % (7).
Le risque cumulé d’adénocarcinome de l’endomètre
est estimé à 50 % à l’âge de 70 ans. Ce risque est plus
élevé en cas de mutation du gène MSH6. Dans un
quart des cas, le cancer de l’endomètre est la première
manifestation tumorale chez les femmes atteintes (7).
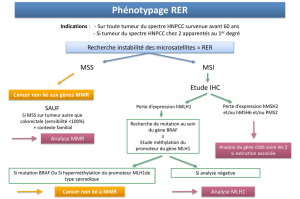
Diagnostic génétique
En cas de suspicion de syndrome HNPCC chez un
patient atteint d’un cancer du spectre du syndrome
HNPCC (cas index), il convient de l’adresser en
consultation d’oncogénétique. Les objectifs de
cette consultation seront d’établir un arbre généa-
logique précis, de valider les diagnostics de cancer,
de demander la recherche d’un phénotype tumoral
MSI+ en biologie moléculaire si celle-ci n’a pas été
réalisée au préalable, et de poser ou non l’indica-
tion d’une analyse génétique constitutionnelle des
gènes MMR. Si le diagnostic de syndrome HNPCC
est retenu, il sera proposé, en collaboration avec les
différents médecins concernés (gastroentérologue,
gynécologue…), une surveillance médicale du patient
et de ses apparentés en attendant l’éventuelle identi-
fication d’une mutation délétère d’un gène MMR chez
le cas index permettant de restreindre la surveillance
aux apparentés porteurs de la mutation délétère.
La recherche d’une mutation constitutionnelle
d’un gène MMR chez un cas index suspect d’être
atteint d’un syndrome HNPCC est parfois longue et
complexe : le délai moyen d’obtention d’un résultat
est compris entre 6 mois et 1 an. Un test diagnos-
tique génétique prédictif chez les apparentés d’un
cas index porteur d’une mutation délétère ne pourra
être proposé qu’à l’âge de début de la mise en œuvre
des mesures de dépistage ou de prévention.
Les critères d’Amsterdam II sont peu sensibles et trop
restrictifs et, appliqués à la lettre, ils ne permettent
pas de reconnaître de nombreuses familles atteintes
d’un syndrome HNPCC. En pratique, une approche
clinico-biologique moins restrictive est utilisée pour
l’identification du syndrome HNPCC, permettant de
sélectionner les patients à adresser en consultation
d’oncogénétique (9). Elle repose sur une stratégie en
deux étapes pour les patients ne remplissant pas les
critères d’Amsterdam. La première étape est basée
sur la recherche d’un phénotype tumoral MSI+ dont
le but est de repérer, parmi les patients atteints d’un
cancer colorectal, le sous-groupe de ceux pouvant
bénéficier d’un diagnostic génétique à la recherche
d’une mutation délétère constitutionnelle d’un
gène MMR. Toutefois, la recherche systématique
chez tous les patients atteints d’un cancer colorectal
avec un phénotype tumoral MSI+ n’est pas réalisable
en pratique, car plus des trois quarts de ces cancers
colorectaux sont de type sporadique. Mais le recours
à quelques critères cliniques simples pour poser l’in-
dication de ce test permet d’améliorer nettement
son efficacité. Afin d’augmenter la sensibilité de
détection des patients porteurs d’une mutation
constitutionnelle délétère d’un gène MMR parmi la
population atteinte de cancer colorectal, d’autres
paramètres, non limités aux critères d’Amsterdam,
et prédictifs de la présence d’un phénotype tumoral
MSI+, ont été proposés. Il s’agit tout d’abord des
critères dits de Bethesda, établis en 1997 puis révisés
en 2004 (tableau II) [10]. La présence d’au moins
un de ces critères est une indication de recherche
Tableau II. Critères de Bethesda révisés.
• Cancer colorectal diagnostiqué avant l’âge de 50 ans.
• Deux cancers synchrones ou métachrones du spectre large du syndrome HNPCC,
quel que soit l’âge de diagnostic des cancers, chez un même patient.
• Cancer colorectal ayant les caractéristiques anatomopathologiques des cancers MSI+
(infiltrat lymphocytaire important, réaction lymphocytaire
Crohn’s-like
, cellule en bagues à chatons,
composante mucineuse importante) à un âge inférieur à 60 ans.
• Cancer colorectal avec au moins un apparenté du premier degré atteint d’un cancer du spectre
du syndrome HNPCC et, dans un cas, un âge au diagnostic de cancer inférieur à 50 ans.
• Cancer colorectal avec au moins deux apparentés du premier ou du deuxième degré
atteints d’un cancer du spectre du syndrome HNPCC, quel que soit l’âge au diagnostic de cancer.

Figure 3. Indications de consultation d’oncogénétique et d’analyse génétique constitu-
tionnelle des gènes MMR.
Cancer (côlon-rectum, utérus, grêle, urothélium, voies biliaires, estomac, ovaire)
Analyse constitutionnelle des gènes MMR
40-60 ans
ou
antécédent*
1er degré
Non muté
Extinction
MSH2/MSH6
IHC : immunohistochimie.
* antécédent de cancer
du spectre du syndrome HNPCC.
muté
< 40 ans
ou antécédent*
personnel
MSI?
Consultation d’oncogénétique
MSI+
IHC recommandée
BRAF (V600E)
Extinction
MLH1
IHC
MSI–
La Lettre du Cancérologue • Vol. XIX - n° 2 - février 2010 | 119
DOSSIER THÉMATIQUE
d’un phénotype tumoral instable MSI+. Plus récem-
ment, l’expertise collective française pour la prise en
charge du syndrome HNPCC a également proposé
un élargissement des critères d’Amsterdam pour la
recherche d’une mutation constitutionnelle délétère
d’un gène MMR (9). En l’absence des critères d’Ams-
terdam élargis, une stratégie en deux étapes, utili-
sant la recherche d’un phénotype tumoral MSI+, est
recommandée (figure 3). Afin de privilégier la sensi-
bilité de la détection des mutations des gènes MMR,
l’indication d’une consultation d’oncogénétique en
vue d’une analyse génétique constitutionnelle des
gènes MMR sera proposée aux patients atteints
d’un cancer du spectre large du syndrome HNPCC
dans les situations suivantes : présence des critères
d’Amsterdam II “élargis” (deux apparentés au premier
degré au minimum et non trois), cancer diagnostiqué
avant l’âge de 40 ans et/ou antécédent personnel
de cancer du spectre large du syndrome HNPCC.
Quant à la recherche d’un phénotype tumoral MSI+,
elle est à demander pour les patients opérés d’un
cancer du côlon avec au moins un des deux critères
suivants : âge au diagnostic entre 40 et 60 ans, anté-
cédent au premier degré de cancer du spectre large
du syndrome HNPCC. En présence d’un phénotype
tumoral MSI+, la recherche de la mutation V600E
du gène BRAF au niveau de la tumeur est utile, car
sa présence signe le caractère sporadique du phéno-
type tumoral MSI+ associé à l’hyperméthylation du
promoteur du gène MLH1 (11). En cas de phénotype
tumoral MSI+ sans mutation V600E du gène BRAF,
l’indication d’une consultation d’oncogénétique sera
retenue pour une analyse génétique constitutionnelle
des gènes MMR. En cas d’impossibilité d’obtenir une
recherche du phénotype tumoral MSI+ dans les cas
ne remplissant pas les critères d’Amsterdam, l’indi-
cation d’une consultation d’oncogénétique en vue
d’une analyse génétique constitutionnelle des gènes
MMR sera proposée si deux apparentés au premier
degré sont atteints d’un cancer du spectre large du
syndrome HNPCC avant 60 ans.
L’immunohistochimie à la recherche d’une extinction
des protéines hMLH1, hMSH2, hPMS2 ou hMSH6
au sein du tissu tumoral est complémentaire de
la recherche d’un phénotype tumoral MSI+ en
biologie moléculaire (12). Le taux de faux positifs
de l’immuno histochimie, c’est-à-dire l’absence
d’extinction d’une de ces quatre protéines alors que
le phénotype tumoral est MSI+, est de 5 à 10 %.
Par conséquent, le diagnostic de syndrome HNPCC
ne peut être formellement rejeté face à l’absence
d’extinction d’une protéine codée par les gènes MMR
en immunohistochimie. Après la détection d’un
phénotype tumoral MSI+ dans le cadre de la stra-
tégie en deux temps, l’immunohistochimie est utile
pour orienter la recherche d’une mutation constitu-
tionnelle sur un gène MMR dont la protéine ne sera
pas exprimée au niveau de la tumeur. Si, d’emblée,
une analyse génétique constitutionnelle des gènes
MMR est proposée, l’étude immunohistochimique
de l’expression des protéines MMR au niveau de la
tumeur est utile pour orienter la recherche d’une
mutation constitutionnelle d’un gène MMR.
Prévention et dépistage
des cancers associés
au syndrome HNPCC
Le risque élevé de cancers associé au syndrome
HNPCC justifie une stratégie de dépistage et de
prévention (tableau III), qui a largement fait la preuve
de son efficacité en termes de réduction de l’inci-
dence du cancer colorectal et du taux de mortalité par
cancer colorectal grâce à la pratique de coloscopies de
dépistage chez les sujets appartenant à des familles
présentant les critères d’Amsterdam (13). Cette stra-
tégie concerne les sujets asymptomatiques dont le
risque génétique a été identifié au niveau individuel
par un diagnostic génétique moléculaire ou dont la
probabilité d’avoir un syndrome HNPCC est élevée.
Chez tous les apparentés d’une personne atteinte,
quand on ne dispose pas d’un diagnostic génétique,
Tableau III. Risques cumulés au
cours de la vie des cancers du
spectre du syndrome HNPCC
chez les patients porteurs
d’une mutation délétère d’un
gène MMR.
Site Risque (%)
Côlon-rectum 80
Endomètre 50
Ovaire 10
Estomac 10
Tractus biliaire 5
Urothélium 5
Grêle 1-5
STOP

120 | La Lettre du Cancérologue • Vol. XIX - n° 2 - février 2010
DOSSIER THÉMATIQUE
Formes héréditaires
des cancers colorectaux Le syndrome HNPCC/Lynch
et chez les seuls porteurs d’une mutation délétère
constitutionnelle, quand on dispose du diagnostic
génétique, la prévention et le dépistage du cancer
colorectal par coloscopie totale doivent débuter
entre 20 et 25 ans ; l’examen doit être répété tous
les 2 ans, si la coloscopie est normale (9). Il convient
d’insister sur la nécessité d’une coloscopie de bonne
qualité, avec une préparation colique parfaite et
le recours systématique à la chromoendoscopie
en utilisant le colorant de surface indigo carmin.
La chromoendoscopie permet de détecter un plus
grand nombre d’adénomes chez les patients atteints
d’un syndrome HNPCC (14). La technique de chro-
moendoscopie sera utilisée après un examen conven-
tionnel de l’ensemble de la muqueuse colorectale.
La surveillance devient annuelle après une colec-
tomie segmentaire pour cancer ou après exérèse
d’un adénome. La réalisation d’une colectomie
prophylactique n’est pas recommandée. Étant
donné le risque de cancer colorectal métachrone,
certaines équipes anglo-saxonnes préconisent la
réalisation systématique d’une colectomie totale
dans le cadre du traitement d’un premier cancer
colorectal chez les patients atteints du syndrome
HNPCC, avec anastomose iléorectale ou iléo-anale
en cas de cancer du rectum. Cette attitude n’est pas
recommandée en France en raison de la morbidité
du geste opératoire et des performances élevées
de la coloscopie de dépistage. Il faut signaler que
l’intérêt de la chimioprophylaxie pour la prévention
des lésions colorectales n’est pas démontré.
En ce qui concerne la surveillance de l’endomètre, un
examen gynécologique et une échographie pelvienne
endovaginale annuelle à partir de l’âge de 30 ans
(8, 9) sont recommandés. L’intérêt de l’hystéroscopie
souple avec biopsies systématiques est en cours
d’évaluation. Contrairement au cancer du côlon,
l’efficacité d’un dépistage du cancer de l’endo-
mètre n’a pas été démontrée. En revanche, l’intérêt
d’une hystérectomie prophylactique a été suggéré
récemment dans une étude rétrospective (15).
Une chirurgie prophylactique du cancer de l’endo-
mètre peut se discuter à partir de l’âge de 40 ans
étant donné l’âge relativement tardif de survenue
de ce cancer, a fortiori en cas d’indication d’une
autre chirurgie pelvienne. Elle peut être réalisée,
par exemple, à l’occasion du traitement chirurgical
d’un cancer du côlon. Compte tenu du risque relatif
augmenté de cancer gastrique associé au syndrome
HNPCC, une surveillance endoscopique œsogas-
troduodénale systématique est recommandée en
cas d’antécédent familial de cancer gastrique, et
il est raisonnable de la proposer à l’occasion des
coloscopies de dépistage sous anesthésie générale,
au minimum dans le but de rechercher Helicobacter
pylori afin de l’éradiquer. En cas d’antécédent fami-
lial de cancer des voies urinaires, certains auteurs
recommandent la recherche annuelle d’une héma-
turie microscopique à partir de l’âge de 40 ans ou
5 ans avant l’âge le plus jeune de survenue de ce
type de cancer au sein de la famille. Pour les autres
localisations tumorales, il n’existe pas de recomman-
dations de surveillance. L’intérêt de la surveillance
du grêle par vidéocapsule a été récemment rapporté
et mérite d’être confirmé.
Conclusion
Le syndrome HNPCC est l’exemple caractéristique
des progrès réalisés dans le domaine de la génétique
du cancer colorectal. D’une part, la compréhension
des altérations génétiques tumorales associées au
syndrome HNPCC a permis d’identifier un méca-
nisme moléculaire de carcinogenèse colorectale
caractérisé par un phénotype tumoral MSI+ dont
la recherche, au moyen de l’analyse en biologie
moléculaire de marqueurs microsatellites, permet
la reconnaissance des cancers se développant dans
le cadre du syndrome HNPCC. D’autre part, l’iden-
tification des gènes responsables de ce syndrome a
permis de disposer d’outils diagnostiques permettant
de dépister des sujets ayant une prédisposition géné-
tique au cancer colorectal avec, comme corollaire,
la mise en place de stratégies de dépistage et de
prévention dont l’efficacité est démontrée. ■
1. Hendriks YM, de Jong AE, Morreau H et al. Diagnostic
approach and management of Lynch syndrome (hereditary
nonpolyposis colorectal carcinoma): a guide for clinicians.
CA Cancer J Clin 2006;56:213-25.
2. Gruber SB. New developments in Lynch syndrome (hereditary
nonpolyposis colorectal cancer) and mismatch repair gene
testing. Gastroenterol 2006;130:577-87.
3. Laghi L, Bianchi P, Malesci A. Differences and evolution of
the methods for the assessment of microsatellite instability.
Oncogene 2008;27:6313-21.
4. Herman JG, Umar A, Polyak K et al. Incidence and func-
tional consequences of hMLH1 promoter hypermethylation in
colorectal carcinoma. Proc Natl Acad Sci USA 1998;95:6870-5.
5. Vasen H, Watson P, Mecklin JP et al. New clinical criteria
for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch
syndrome) proposed by the International Collaborative Group
on HNPCC. Gastroenterol 1999;116:1453-8.
6. Hampel H, Stephens JA, Pukkala E et al. Cancer risk in here-
ditary nonpolyposis colorectal cancer syndrome: later age of
onset. Gastroenterol 2005;129:415-21.
7. Parc Y, Boisson C, Thomas G, Olschwang S. Cancer risk
in 348 French MSH2 or MLH1 gene carriers. J Med Genet
2003;40:208-13.
8. Koornstra JJ, Mourits MJ, Sijmons RH et al. Management of
extracolonic tumours in patients with Lynch syndrome. Lancet
Oncol 2009;10:400-8.
9. Olschwang S, Bonaiti C, Feingold J et al. Identification and
management of HNPCC syndrome (hereditary non polyposis
colon cancer), hereditary predisposition to colorectal and
endometrial adenocarcinomas. Bull Cancer 2004;91:303-15.
10. Umar A, Boland CR, Terdiman JP et al. Revised Bethesda
guidelines for hereditary nonpolyposis colorectal cancer (Lynch
syndrome) and microsatellite instability. J Natl Cancer Inst
2004;96:261-8.
11. Olschwang S, Paraf F, Laurent-Puig P et al. [Recent advances
for the identification and screening of Lynch syndrome]. GCB
2007;31:136-40.
12. Hampel H, Frankel WL, Martin E et al. Feasibility of screening
for Lynch syndrome among patients with colorectal cancer.
JCO 2008;26:5783-8.
13. De Jong AE, Hendriks YMC, Kleibeuker JH et al. Decrease in
mortality in Lynch syndrome families because of surveillance.
Gastroenterol 2006;130:665-71.
14. Lecomte T, Cellier C, Meatchi T et al. Chromoendoscopic
colonoscopy for detecting preneoplastic lesions in hereditary
nonpolyposis colorectal cancer syndrome. Clin Gastroenterol
Hepatol 2005;3:897-902.
15. Schmeler KM, Lynch HT, Chen LM et al. Prophylactic surgery
to reduce the risk of gynecologic cancers in the Lynch syndrome.
NEJM 2006;354:261-9.
Références bibliographiques
1
/
5
100%