Les nouvelles cibles thérapeutiques. Les nouvelles thérapeutiques

Ces dernières années, des avancées importantes ont été réalisées dans le domaine de
la recherche fondamentale et de transfert concernant les mécanismes impliqués
dans les processus d’acquisition d’un phénotype cancéreux par une cellule normale
du sein. Ces nouvelles acquisitions ont concerné la transduction du signal, le cycle
cellulaire, l’apoptose, l’angiogenèse, la migration et l’invasion cellulaire. Des nou-
velles cibles thérapeutiques potentielles ont ainsi été identifiées et de nouvelles thé-
rapeutiques ciblées sont de ce fait développées par des institutions académiques, des
sociétés de biotechnologie et des compagnies pharmaceutiques. Certains de ces
agents agissant sur des nouvelles cibles thérapeutiques sont déjà utilisés en clinique,
d’autres sont en évaluation. Nous nous intéresserons essentiellement aux nouveaux
agents intervenant au niveau de la transduction du signal. L’hormonothérapie des
cancers du sein étant une thérapeutique ciblée au niveau des récepteurs hormonaux
estrogéniques, nous évoquerons les échanges « cross talks » entre cette voie classique
et la transduction du signal.
Le passage d’une cellule eucaryote de la phase quiescente (G0) à la phase de
mitose (M), en réponse à des stimuli extérieurs est un processus multi-étape.
Il requiert la transduction de signaux divers (prolifération, migration, différencia-
tion) et l’activation de protéines intracellulaires. Ce processus finement régulé est
altéré au sein des cellules néoplasiques, engendrant une prolifération, une migration
et une différenciation cellulaire non contrôlée.
Cette communication inter- et intra-cellulaire peut être schématiquement
divisée en trois étapes. La première étape consiste en la fixation sur un récepteur
trans-membranaire d’un signal extracellulaire. La deuxième étape est l’activation de
seconds messagers intracellulaires aboutissant à l’activation de facteurs de transcrip-
tions qui vont pouvoir agir directement sur la transcription de gènes impliqués dans
les processus de prolifération, de migration et de différenciation cellulaire. La troi-
sième étape correspond au cycle cellulaire.
Les nouvelles cibles thérapeutiques.
Les nouvelles thérapeutiques ciblées
P. Fumoleau, M. Campone, N. Isambert, E. Bourbouloux, F. Mayer
et B. Coudert

Les récepteurs trans-membranaires, des cibles thérapeutiques
Sont actuellement décrites six familles (tableau 1) de récepteurs tyrosine kinase qui
se différencient par leur ligand et leur structure chimique, mais qui possèdent des
caractéristiques communes, et sept récepteurs « orphelins » (1) :
– un domaine extra-cellulaire, site de fixation du ligand (partie N terminale de la
molécule) ;
– un domaine transmembranaire, site d’ancrage dans la membrane cytoplasmique ;
– un domaine intracellulaire riche en résidu tyrosine, possédant une activité tyro-
sine kinase.
Tableau 1 - Récepteurs tyrosine kinase.
Famille Classe I : Classe II : Classe III : Classe IV : Classe V : Classe VI :
de récepteur ErbB FGF HGF insuline neurotrophines PDGF
Ligand EGF, TGF, FGF 1 à 8 HGF Insuline, NGF PDGF-A et B
AR, HB-EGF, IGF-1 BDNF VEGF
SDGF, IGF-2 NT-3 à 5 CSF-1
Hereguline, SCF
Bêtacelluline PIGF
Récepteur HER-1 FGFR-1 MET IR TRKA PDGFR alpha
HER-2 FGFR-2 IGFI-R TRKB et bêta
HER-3 FGFR-3 TRKC CSF-1R
HER-4 FGFR-4 SCFR
FLT
KDR
234 Cancer du sein

La famille ErbB
Elle comprend quatre récepteurs tyrosine kinase, ErbB-1 (aussi appelé epidermal
growth factor receptor [EGFR] ou HER1), ErbB-2 (mieux connu sous le nom de
HER2/neu), ErbB-3 (HER3), et ErbB-4 (HER4).
Les récepteurs ErbB
Ils ont un rôle important au niveau de la croissance normale et le développement.
Il sont associés à des processus divers comme la division cellulaire, la survie, l’angio-
genèse, la motilité et l’adhésion (2). Une altération du signal au niveau de ces récep-
teurs ErbB peut ainsi déstabiliser ces processus et contribuer à la transformation
maligne (3). La relation entre les récepteurs ErbB et le cancer a été rapporté dans les
années 1980 quand il a été découvert que l’oncogène contenu dans la tumeur éry-
throblastique aviaire codait pour une forme mutée de ErbB-1/EGFR
(v-erbB) (3, 4). Depuis, de nombreuses études ont démontré une association entre
des taux élevés ou des formes mutées des récepteurs ErbB et de nombreuses
tumeurs (2, 5). De plus, la présence d’un récepteur ErbB hyper exprimé ou muté est
associé à un mauvais pronostic et/ou à une diminution de la réponse à la chimio-
thérapie dans beaucoup de tumeurs (2, 5). Ainsi, dans le cancer du sein, une hyper-
expression de ErbB-1 est retrouvée dans 14 à 91% des études et, pour ErbB-2, dans
10 à 37 %. Les récepteurs ErbB hyperexprimés ou mutés sont toujours capables de
répondre à des stimuli extérieurs. Cet élément fait qu’ils sont des candidats pour le
développement des agents thérapeutiques ciblés.
L’interaction avec un ligand
Elle induit une dimérisation du récepteur, élément critique pour l’initiation du
signal intracellulaire. Les récepteurs dimérisés sont alors activés via à la fois une
autophosphorylation et une transphosphorylation transmoléculaire des résidus clés
de type tyrosine kinase au niveau des domaines cytoplasmiques (5). Ces phospho-
rylations des résidus tyrosines servent de sites de liaisons pour d’autres molécules
d’amont intervenant dans la transduction du signal à travers des kinases addition-
nelles. Des molécules adaptatrices possédant un domaine d’homologie Src (SH2
domaine) et un domaine de fixation à tyrosine sont à leur tour activées. Deux
grandes voies de la transduction du signal sont privilégiées : la voie Ras/Raf/MAKK-
MEK/ERK, la voie des phospho-inositol/PI3Kinase/AKT, mais aussi les voies pas-
sant par la phospholipase C et la voie STATS (PAK-JNKK-JNK). Elles vont induire
la phosphorylation de facteurs de transcription, Jun, Fos, myc, cycline D1, induisant
la transcription de protéines impliquées dans les mécanismes de prolifération cellu-
laire, d’angiogenèse, de migration, de différenciation cellulaire ou bien encore dans
la survie cellulaire.
Les paires de récepteurs identiques sont appelées homodimères, alors que les
paires composées de différents récepteurs sont des hétérodimères. Dans la famille
ErbB, les deux types de combinaison sont possibles. Mais il semble exister une hié-
Les nouvelles cibles thérapeutiques. Les nouvelles thérapeutiques… 235

rarchie bien définie pour la formation de ces dimères. Ainsi, ErbB-2 qui n’apparaît
pas avoir de ligand est le partenaire favori de dimérisation des autres récepteurs
ErbB (7). Les hétérodimères contenant ErbB-2 ont des caractéristiques particu-
lières, comme une dissociation avec le ligand et une endocytose plus lentes, indui-
sant un signal d’amont plus prolongé et plus puissant permettant une prolifération
cellulaire plus importante (3).
Homo- et hétérodimères
Voie d’activation de signalisation ErbB
236 Cancer du sein

L’activation des récepteurs ErbB
Elle induit aussi l’internalisation cytoplasmique du récepteur via un processus
incluant l’endocytose. Les récepteurs internalisés sont, soit dégradés dans les com-
partiments endosomales, soit recyclés avec retour au niveau de la surface cellulaire.
Ces phénomènes dépendent de la composition des dimères. Les homo- ou hétéro-
dimères contenant ErbB-1 sont le plus souvent dégradés, ceux contenant ErbB-3
sont recyclés et ceux contenant ErbB-2 bénéficient d’une endocytose plus lente et
d’un recyclage plus important au niveau de la surface cellulaire. Ces processus
impliquent l’ubiquitine ligase c-Cbl qui induit une poly-ubiquitination des homodi-
mères ErbB1 orientés ensuite vers une dégradation lysosomale.
L’activation du signal à partir de ErbB
Elle est retrouvée dans plusieurs types tumoraux, en particulier dans le cancer du
sein. Différents mécanismes peuvent contribuer à une dysrégulation incluant une
hyperexpression des récepteurs et/ou des ligands et des mutations géniques indui-
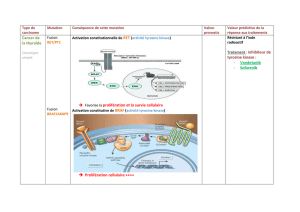
sant des récepteurs constitutivement actifs. Concernant ErbB-1, la mutation la plus
fréquente est EGFRvIII (8). Cette mutation est induite par une délétion des exons
2 à 7 aboutissant à une réduction du domaine extracellulaire (8, 5). Ce récepteur ne
peu plus se lié au ligand, cependant cette mutation induit l’activation constitutive
du domaine kinase, ce qui peut contribuer à la transformation cancéreuse.
EGFRvIII n’est pas exprimé par les cellules normales, mais retrouvé dans différents
types tumoraux comme les gliomes et certains cancers du sein, de la prostate et du
poumon non à petites cellules (9, 5).
La dysrégulation peut aussi se produire à travers l’hyperexpression du récepteur
et/ou du ligand. Des études in vitro suggèrent que, dans le cas d’ErbB-1, l’expression
du ligand est nécessaire pour la transformation induite par l’hyperexpression du
récepteur non muté. Une co-hyperexpression d’ErbB-1 et de ses ligands, particuliè-
rement EGF et TGF-α, est fréquemment retrouvée au niveau des tumeurs primi-
tives, créant ainsi une boucle autocrine pour la croissance tumorale (10).
À l’inverse, l’hyper-expression du récepteur non muté ErbB-2 permet une acti-
vation des voies de signalisation indépendamment du ligand. L’hyperexpression de
ErbB-2 est une caractéristique de 20 à 25 % des cancers du sein (11).
ErbB-2
Il est exprimé sous la forme d’une protéine trans-membranaire de 185-kD qui peut
faire l’objet d’un clivage protéolytique à la surface cellulaire et aboutissant au relar-
gage (shedding) d’un fragment ECD (extra cellular domain) et à la persistance d’un
fragment de 95-kD contenant les domaines transmembranaires et cytoplasmiques
(12,13). Il a été montré que ce fragment 95-kD gardait une activité kinase in vitro
(13, 14), suggérant qu’il pouvait être constitutivement actif avec augmentation
in vivo du potentiel de transformation. La présence du fragment p95 dans de nom-
breuses tumeurs primitives mammaires est corrélée avec l’importance de l’envahis-
sement ganglionnaire (13, 15). De plus, le taux d’ECD circulant semble être
Les nouvelles cibles thérapeutiques. Les nouvelles thérapeutiques… 237
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
1
/
34
100%