Introduction à l`oncogénétique

UE1 – Cours n°24 – Dr B. DEMEER – 10/04/13 Typ: Junélie / Cor: Yann
Introduction à l’oncogénétique
1- Introduction à la prédisposition génétique aux cancers.
A) Historique.
Le premier gène de prédisposition héréditaire au cancer a été découvert en 1986, c’est le gène Rb du
rétinoblastome. Depuis les années 1990 d’autres gènes de prédisposition ont été découverts. Depuis ce temps est
née l’oncogénétique, ainsi que des recommandations concernant la surveillance et la prise en charge.
On décrit des syndromes génétiques, associant à la fois des anomalies du développement et une prédisposition aux
cancers.
On décrit également une prédisposition monogénique aux cancers.
B) Exemples de pathologies entrainant une prédisposition génétique à divers cancers.
Par exemple, la trisomie 21 associe un syndrome dysmorphique, un syndrome malformatif, un retard mental et des
prédispositions aux infections, aux pathologies auto-immunes et à la leucémie (risque relatif multiplié par 20).
Dans le syndrome de Klinefelter (caryotype 47,XXY) on retrouve une grande taille, une infertilité et une
prédisposition au cancer du sein.
Dans le syndrome WAGR, due à une anomalie chromosomique à type de micro délétion 11p14, on retrouve
l’association d’une tumeur de Wilms (néphroblastome), d’une aniridie, d’anomalies génito-urinaires et d’un retard
mental.
Dans le syndrome de Gorlin on retrouve une naevomatose basocellulaire (carcinome basocellulaire, kératokystes
odontogéniques), il est lié au gène PTCH (pénétrance complète, expressivité variable).
Dans le syndrome de Cowden on retrouve des hamartomes multiples et un risque de développer des tumeurs
malignes ; il est lié au gène PTEN.
C) Agrégation familiale.
Cancers colorectaux (syndrome de Lynch).
Les carrés correspondent aux hommes, les ronds aux femmes. La couleur blanche
signifie que le patient est sain, la couleur noire que le patient est malade, le fait
que le carré ou le rond soit barré signifie que le patient est décédé. La présence
d’une étoile signifie que le patient possède une mutation génétique.
CCR signifie cancer colo rectal et le chiffre qui le suit correspond à l’âge (exemple :
CCR 64 signifie que le patient a développé un cancer colorectal a 64 ans).
La prédisposition génétique entraine l’apparition de cancers chez des sujets jeunes par rapport aux normes
d’apparition.
Cancers du sein et de l’ovaire.

UE1 – Cours n°24 – Dr B. DEMEER – 10/04/13 Typ: Junélie / Cor: Yann
La prédisposition génétique n’est recherchée que d’un côté de l’arbre, par exemple si l’on prend le côté gauche de
l’arbre on observe que sur 5 femmes 3 ont été atteintes du cancer du sein.
2- De la biologie des cancers à la prédisposition génétique aux cancers.
A) La transformation tumorale.
On retrouve une accumulation progressive des mutations au cours des divisions cellulaires. L’expansion clonale
débute quand un gène de régulation de la prolifération cellulaire ou de réparation de l’ADN est muté.
Lorsque l’on retrouve un équilibre entre les facteurs proto-oncogènes et les gènes suppresseurs de tumeurs, le
système est homéostasique.
En cas d’activation des proto-oncogènes et d’inhibition des gènes suppresseurs de tumeur, on retrouve une
prolifération anarchique.
Le phénomène cancéreux débute ainsi par une mutation, entrainant une compétition et finalement la sélection. Les
cellules transformées sélectionnées se divisent indépendamment des autres cellules voisines.
On retrouve une augmentation de l’activité des oncogènes (gènes favorisant la croissance tumorale) et une
inactivation des gènes suppresseurs de tumeur (gènes s’opposant à la transformation tumorale).
NB : passenger mutation sont les mutations subies physiologiquement au fil du temps par les cellules, driver mutation
sont les mutations à l’origine d’une prolifération anarchique aboutissant à un mutator phenotype.

UE1 – Cours n°24 – Dr B. DEMEER – 10/04/13 Typ: Junélie / Cor: Yann
Ainsi le cancer est composé de cellules transformées au sein d’un tissu normal de l’organisme, caractérisées par la
perte des caractéristiques de la cellule eucaryote normale (dérégulation du cycle cellulaire, altération des voies de
signalisation cellulaires, défaut des systèmes de réparation de l’ADN) et l’acquisition de caractéristiques spéciales :
- Indépendance aux facteurs de croissance.
- Insensibilité aux signaux antiprolifératifs.
- Echappement à l’apoptose.
- Réplication illimitée.
- Induction de l’angiogenèse.
- Invasion tissulaire et capacité à métastaser.
- Instabilité génétique.
B) L’apparition du cancer.
Les rayonnements UV, les irradiations, les agents chimiques (hydrocarbures), les agents infectieux (VHB, VHC, HPV),
les facteurs génétiques (syndrome de prédisposition génétique) sont tous des agents carcinogènes.
Ces carcinogènes entrainent des anomalies génétiques (initiation et promotion).
Le cancer est différent d’une simple maladie génétique car il n’est pas lié à la mutation d’un unique gène donné mais
à l’accumulation de mutations de plusieurs gènes distincts.
Le nombre d’étapes minimales varie avec le type de cancer. Par exemple dans le cancer du côlon on retrouve une
inactivation de APC et donc une activation de la β caténine ce qui entraine le passage de l’épithélium normal à la
crypte anormale. L’activation de K-Ras entraine le passage à l’adénome précoce, puis l’inactivation de SMAD 4
aboutit à l’adénome tardif. Suite à cela l’inactivation de la p53 aboutit au développement du carcinome, et
finalement l’inactivation de la E-cadhérine aboutit au carcinome invasif.
C) Prédisposition aux cancers.
Définition de la prédisposition génétique.
On retrouve la notion de prédisposition génétique aux cancers, c’est-à-dire une augmentation du risque de cancer
d’une personne par rapport au risque de la population générale.
Au niveau biologique la prédisposition génétique correspond à une situation où le quota de mutation nécessaire à la
transformation cellulaire est atteint plus rapidement que dans la population générale.
Les gènes concernés sont les oncogènes et les gènes suppresseurs de tumeurs (gènes régulant le cycle cellulaire ou
gatekeepers, et les gènes de maintenance et de réparation de l’ADN ou caretakers).
Mode de transmission et caractéristiques.

UE1 – Cours n°24 – Dr B. DEMEER – 10/04/13 Typ: Junélie / Cor: Yann
Le mode de transmission est le plus souvent autosomique dominant (la présence d’un seul allèle muté est suffisante
pour que la maladie s’exprime). On parle de mutation constitutionnelle en opposition à une mutation somatique
normale au cours du développement.
Les deux sexes sont souvent atteints avec la même fréquence. La transmission de la maladie peut se faire par les
deux sexes, il existe en particulier une transmission père-fils.
Tout sujet porteur d’un allèle morbide dominant a 50% de chance de le transmettre à chacun de ses enfants, quel
que soit le sexe.
Les sujets sont donc atteints sur plusieurs générations, ce qui donne une répartition verticale sur l’arbre
généalogique.
- Pénétrance incomplète.
Pour certaines maladies, un individu connu porteur de la mutation (soit par sa position sur l’arbre généalogique soit
par l’analyse moléculaire) peut ne présenter aucun signe de l’affection. Le gène morbide est dit alors avoir une
pénétrance incomplète.
Un sujet apparemment sain peut donc être porteur du gène muté et transmettre la maladie à sa descendance
donnant lieu ainsi à un « saut de génération ».
La pénétrance d’un gène morbide peut aussi varier en fonction d’autres caractères comme l’âge ou le sexe.
- Expressivité variable.
L’expression d’un allèle morbide est différente d’un individu à l’autre.
- Phénocopie.
La phénocopie est définie par l’existence du même cancer chez une personne de la famille non porteuse de la
prédisposition familiale.
D) Exemple du rétinoblastome.
Le rétinoblastome est une tumeur rare de l’œil, l’incidence de ce cancer est de 1/15 000.

UE1 – Cours n°24 – Dr B. DEMEER – 10/04/13 Typ: Junélie / Cor: Yann
risque
Dans 60% des cas il s’exprime par une tumeur unilatérale après 12 mois, dans 40% des cas il s’exprime par une
tumeur bilatérale de moins de 12 mois, et dans 10% des cas de tumeur bilatérale précoce surtout un des deux
parents a été atteint dans l’enfance (prédisposition génétique).
L’hypothèse de Knudson et Comings décrit les faits suivants :
- Dans la forme isolée tardive il faut deux mutations dans le même gène de la cellule pour entrainer le
rétinoblastome.
- Dans la forme familiale une mutation est déjà présente dans toutes les cellules car héritée, il existe donc plus
de chances de voir apparaitre une deuxième mutation ce qui explique l’atteinte plus précoce et surtout
bilatérale.
Cette hypothèse a été confirmée par la découverte du gène Rb1 qui est un gène suppresseur de tumeur. Dans une
cellule saine homozygote, une mutation ponctuelle transforme la cellule homozygote en cellule hétérozygote ;
l’accumulation de mutations du gène Rb1 dans la même cellule permet de retrouver une homozygotie
(pathologique) ce qui entraine la prolifération tumorale.
En cas de prédisposition génétique, la cellule-mère est déjà du départ hétérozygote.
3- Généralités sur l’oncogénétique.
A) Généralités.
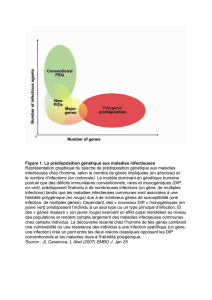
Actuellement 76 gènes ont été identifiés. La prédisposition subit une transmission selon un mode mendelien, elle est
associée à un risque tumoral élevé.
Tous les gènes découverts ne sont pas à l’origine d’une prédisposition génétique
au cancer.
Certains gènes (ALK, SUFU, RET, APC, Rb1, CDH1) donnent dans 100% des cas un
cancer dès qu’ils sont mutés. On parle alors de déterminisme monogénique.
 6
7
8
9
10
11
12
6
7
8
9
10
11
12
1
/
12
100%